Content from Introduction
Last updated on 2025-12-12 | Edit this page
Overview
Questions
- What exactly is job efficiency in the computing world?
- Why would I care about job efficiency and what are potential pitfalls?
- How can I start measuring how my program performs?
Objectives
After completing this episode, participants should be able to …
- Use timing commands provided by
timeanddate. - Understand the benefits of efficient jobs in terms of runtime and numerical accuracy.
- Have developed some awareness about the overall high energy consumption of HPC.
Background
Job efficiency, as defined by Oxford’s English Dictionaries, is the ratio of the useful work performed by a machine […] to the total energy expended or heat taken in. In a high-performance-computing (HPC) context, the useful work is the entirety of all calculations to be performed by our (heat-generating) computers. Doing this efficiently thus translates to maximizing the calculations completed in some limited time span while minimizing the heat output. In more extreme words, we want to avoid running big computers for nothing but hot air.
One may object that a single user’s job may hardly have an effect on an HPC system’s power usage since such systems are in power-on state 24/7 anyway. The same may be argued about air travel. The plane will take off anyway, whether I board the plane or not. However, we indeed have some leverage in contributing to efficiency, defined by fuel consumption in air travel: traveling lightly, i.e., avoiding excessive baggage will improve the airplane’s ratio \(\frac{useful\;work}{total\;energy\;expended}\). So let’s get back to the ground and look at some inefficiencies in computing jobs, while we will continue to use the air-travel analogy.
time to sleep
Let’s look at the commandsleep
This command triggers a “computer nap”. It actually delays whatever
would come next for the specified time, here 2 seconds. You can verify
that nap time using a stopwatch, the latter given by
thetimecommand:
which will report something like
real 0m2.002s
user 0m0.001s
sys 0m0.000sThe time command shall be our first
performance-measuring tool. timehas become a bit of a
hello-world equivalent in HPC contexts. This command gives you
a breakdown of how your program uses CPU (Central Processing Unit) and
wall-clock time. The standard output of time reports three
fields, real, user and sys:
| Time | Meaning |
| real | Wall-clock time = total runtime as seen on a stopwatch |
| user | Time spent in user-mode: actual computations like math, loops, logic |
| sys | Time spent in OS’s kernel-mode (system calls): I/O = reading/writing files, handling memory, talking to other devices |
The abovesleepcommand abstains from any kind of math,
I/O, or other work that would show up in user or sys
time, hence these entries show (almost) zero.
Thetimecommand is both a keyword directly built into the
Bash shell as well as an executable file, usually residing
under/usr/bin/time. While very similar, they are not
exactly the same. Shell/Bash keywords take precedence, so preceding a
command withtimeinvokes the shell keyword. Therefore, if
you want to force the usage of/usr/bin/time, you would
do
BASH
# Explicitly calling the `time` binary
$ /usr/bin/time sleep 2
0.00user 0.00system 0:02.00elapsed 0%CPU (0avgtext+0avgdata 2176maxresident)k
0inputs+0outputs (0major+90minor)pagefaults 0swaps
# Compare the output to the Bash built-in:
$ time sleep 2
real 0m2,003s
user 0m0,001s
sys 0m0,003s
# Yet another output of `time` in zsh, an alternative shell implementation to bash
$ time sleep 2
sleep 2 0,00s user 0,00s system 0% cpu 2,003 totalNotice the different output formatting. All tools provide similar insight, but the formatting and exact information may differ. So, if you saw something that looks different from the bash built-in command, this may be why!
Further note, that shell keyword documentation is invoked
viahelp <KEYWORD>, for example
help time, while most executables have manual pages,
e.g.,man time. At last, you can prefix the shell keyword
with a backslash in order to stop Bash from evaluating it,
so\time sleep 2will revert
to/usr/bin/time.
Time for a date
Thedatecommand, as its manpage (man date)
says, prints or sets the system date and time. In fact, this gives us a
super accurate stopwatch when used like this:
reports a point in time as a number of seconds elapsed since a fixed reference point.
A referenced time point is also referred to as Epoch time, where,
according to the manpage ofdate, the (default) reference
point is the beginning of the year 1970, given as “1970-01-01 00:00
UTC”.
While%sinvokes output of a referenced time, the
additional specifier%Nenforces an accuracy down to
nanoseconds. Give it a try and you will see a large number (of seconds)
followed by 9 digits after the decimal point.
An accurate stopwatch: date
You can use the constructdate +%s.%Non the command line
or in a Bash script to save start and end time points as a variable:
This gives you a stopwatch by setting a start time, running some
command(s), and then storing the end time after
command(s) into a second variable. Differencing the two
times produces the elapsed time. Give this a try with
thesleepcommand in between.
Part 1: Example for an inefficient job
After warming up with some timing methods, let’s analyze the
efficiency of a small script that makes our computer sweat a bit more
than thesleepcommand. Have a look at the following Bash
shell 7-liner.
BASH
#!/bin/bash
sum=0
for i in $(seq 1 1000); do
val=`echo "e(2 * l(${i}))" | bc -l`
sum=$(echo "$sum + $val" | bc -l)
done
echo Sum=$sumCopy-paste this to a file, saysum.bash, and make it
executable via
The main part of this shell script consists of
aforstatement which is calculating the sum of all squares
\(i^2\) for 1000 iterations; note
thatseq 1 1000creates the number sequence (\(i=1,2,3,...,1000\)). Inside the
forloop thebccalculator tool is employed. The
first statement inside the loop (val=...) prints the
expression e(2 * l(${i})), which is bc-talk
for the expression \(i^2\) because of
the relation \(i^x=e^{x\cdot \ln(i)}\),
for example \(e^{2\cdot\ln(3)}=3^2\),
where ln is the natural logarithm. The second statement inside the loop
(sum=...) accumulates the expressions
val=\(i^2\)
intosum, so the output of the finalecholine is
the total, \(\sum_{i=1}^{1000}i^2\).
Identify the inefficient pieces
In the above Bash script, theforloop invokes the
bccalculator twice during every loop iteration. Compared to
another method to be investigated below, this method is rather slow. Any
idea why that is the case?
Each statementecho … | bc -lspawns a
newbcprocess via a subshell.
The statementecho … | bc -lspawns a
newbcprocess via a subshell. Here, each loop iteration
invokes two of those. Each subshell is essentially a separate process
and involves a certain startup cost, parsing overhead, and OS-internal
inter‑process communication. Such overhead will account for most of the
total runtime ofsum.bash.
The overhead in this shell script is dominated by process creation
and context switching, that is, calling thebctool so many
times. Going back to our air-travel analogy, the summation of 1000
numbers shall be equivalent to having a total of 1000 passengers board a
large plane. When total boarding time counts, an inefficient boarding
procedure would involve every passenger loading two carryon pieces. Many
of you may have experienced, how stuffing an excessive number of baggage
pieces into the overhead compartments can slow things down in the
plane’s aisles, similar to the overhead due to the 2000 (two for each
loop iteration)bcsub-processes that hinder the data stream
inside the CPU’s “aisles”.
Let’s pull out our stopwatches
Using eithertimeordate, can you get a
runtime measurement forsum.bash?
You can precede any command withtime. If you want to
usedate, remember thatnow=$(date +%s.%N)lets
you store the current time point and&&lets you join
commands together.
A straightforward way is
Alternatively,dateand&&can be
combined to a wrapper in order to
timesum.bashexternally,
Another option is to placedateinside the
scriptsum.bash,
Speeding things up
A remedy to the inefficiencies we found inside
theforloop ofsum.bashis to avoid the spawning
of many sub-processes that are caused by repetitively
callingbc. In other words, ideally, the many sub-processes
conflate into one. In terms of the airplane analogy, we want people to
store all their carryon pieces in a big container, where its subsequent
loading onto the plane is a single process, as opposed to every
passenger running a proprietary sub-process. Collapsing things into one
sub-processes can be achieved by replacing the external loop by
abc-internal one:
In this method, to be called the one-liner, the loop, arithmetic, and accumulation are free of the overhead. This example shall be a placeholder for a common scenario, where potentially large efficiency gains can be achieved by replacing inefficient math implementations by numerically optimized software libraries.
Evaluate the runtime improvement
Compare the runtimes of the summation
scriptsum.bashversus the one-liner.
The Bash keywordtimeis sufficient to see the runtime
difference.
While it depends a bit on the employed hardware, one will notice that
the one-liner runs roughly 1000 times faster thansum.bash.
Of course, one could live with this inefficiency when it is just needed
once in a while and the script’s overall runtime amounts to just a few
seconds. However, imagine some large-scale computing job that is
supposed to finish within an hour on a supercomputer for which one has
to pay a usage fee on a per-hour basis. If implemented poorly, an
already small overhead increase, say by a factor of 2, would render this
computing job expensive, both in terms of time and money.
The above runtime comparisons merely look at calculation speed, which depends on CPU processing speed. Such a task is thus called CPU-bound. On the other hand, the performance of a memory-bound process is limited by the speed of memory access. This happens when the CPU spends most of its time waiting for data to be fetched from memory (RAM), cache, or storage, causing its execution pipeline to stall. Optimization of memory-bound tasks addresses performance bottlenecks due to data transfer speeds rather than calculation speeds. Finally, when data transfer involves a high percentage of disk or network access, disk/networking speed becomes a limiting factor, rendering a process I/O-bound.
To be precise: Numerical efficiency
Inefficient computing is not only limited to being unnecessarily slow. It can also entail the scenario where an excessive accuracy can lead to unnecessary runtime increases. Without going into details, let’s just keep in mind that in computing, accuracy depends on the precision of the numbers that are being processed by the CPU. Precision essentially governs how many digits after the decimal point are accounted for in mathematical operations. The higher the precision, the fewer calculations can be processed within a fixed time. On the other hand, within that same time, the CPU can crank through more low-precision numbers; however, an insufficient precision can render lengthy calculations useless. The optimal degree of precision, in terms of computing efficiency, is application dependent.
Compare numerical results
Our summation implementation viasum.bashexemplifies the
case of an inaccurate calculation. When running the two summation
methods in the previous challenge, have a look at the actual summation
results. Which of the two end results do you think is more accurate and
why? Is the erroneous result smaller or larger and why?
Think of another airplane example. Which scenario is more prone to things getting lost or forgotten? 1) Passengers bring and take their own baggage pieces to the cabin, or 2) Baggage pieces are stored and retrieved collectively.
The methodsum.bash, using the
externalforloop, and the one-liner return the final sums,
respectively,
103075329 # bc, external loop (sum.bash)
333833500 # bc, internal loop (one-liner)where the first result may vary on your machine. The
methodsum.bashis affected by the setting of the
bc-internal parameterscalewhich defines how
some operations, here the exponential function e(…) and
logarithm ln(…) use digits after the decimal point. The
default value ofscaleis 0, which basically leads to
truncations after the decimal point, so rounding errors accumulate at
every loop iteration. Hence, the final sum drifts downward (by a lot)
compared to the second (true) value. Of course,scalecan be
increased. The manpage ofbcactually says that it is “an
arbitrary precision calculator language”.
Part 2: About HPC power consumption
The HP (high performace) in HPC refers to the fact that the employed computer hardware is able to do a lot of multitasking, also called parallel computing. Parallel programming essentially exploits the CPU’s multitasking ability. Therefore, a lot of HPC-efficiency aspects revolve around keeping everyone in a CPU’s multitasking team equally busy. We will look at some of those aspects during the course of later episodes.
The more the merrier: CPU/GPU cores
Common parallel-computing jobs employ multiple cores of a CPU, or even multiple CPUs, simultaneously. A core is a processing unit within a CPU that independently executes instructions. These days (as of 2025), typical CPUs are quad-core (4 cores), octa-core (8 cores), and so on. High-end gaming CPUs often have 16+ cores, HPC cluster nodes feature multiple CPUs, oftentimes with 64+ cores each; and all these numbers keep going up.
Nowadays, almost all HPC centers are also equipped with GPU (Graphics
Processing Unit) hardware. Such hardware is optimal for jobs where
many cores is more important than fewer powerful cores.
The number of GPU cores varies greatly depending on the model, ranging
from a few hundred in low-end GPU cards to over 16,000 in high-end
ones.
Measuring parallel runtime: core hours
Owing to the inherent parallelism in the HPC world, people came up with some measure which takes the granularity into account when allocating not only runtime but also the number of requested cores. The unit core hour (core-h) represents the usage of one CPU core for one hour and scales with core count. For example, assume you have a monthly allocation of 500 core-h, with a fee incurred when exceeding that quota. So with 500 core-h, you could run a one-hour parallel job utilizing 500 CPU cores for free. Or, in the other extreme, if your program does not or cannot multitask, you could run a single-core job for 500 hours, provided you won’t forget at the end what this job was about.
So far, the focus has been on core number and hours for HPC resource allocation. Keep in mind, however, that the HPC resource portfolio involves other hardware components as well:
- Memory: There are (whether parallel or not) jobs, that request a large amount of memory (RAM). For example, some mathematical solution methods for large equation systems do not allow the compartmentalization of the total required memory across CPU cores, that is, many-core processes need to know each other’s memory chunks. HPC centers usually have large-memory nodes assigned for such applications.
- Storage: Other applications process huge amounts of data, think of genomics or climate modelling, which can involve terabytes or even petabytes of data to be stored and analyzed.
A typical HPC computing job
Like in the automotive world, high performance means high power, which in turn involves a high energy demand. Let’s consider a typical parallel scientific-computing job to be run in some HPC center. Our example job shall be deemed too large for one CPU, so it employs multiple CPUs, which in turn are distributed across nodes. Node power usage is measured in W=Watt, which is the SI unit of power and corresponds to the rate of consumption of energy in an electric circuit. One compute node with a 64-core CPU can consume between 300 W in idle state, and 900 W (maximum load) for air-cooled systems, whereas this range is roughly 250-850 W for the slightly more efficient liquid-cooled systems. For comparison, an average coffee maker consumes between 800 W (drip coffee maker) and 1500 W (espresso machine). Our computing job shall then use these resources:
- 12 nodes are crunching numbers in parallel
- 64 cores/node (e.g., Intel® Xeon® 6774P, or AMD® EPYC® 9534)
- 12 hours of full load (realistic for many scientific simulations)
- Power per node: (idle vs. full load):
- Idle: ~300 W
- Full load: ~900 W
- Extra power per node: 600 W
- Total extra power: 12 nodes × 600 W × 12 hours = 86,400 Wh = 86.4 kWh
How many core hours does this job involve?
HPC centers have different job queues for different kinds of computing jobs. For example, a queue named big-jobs may be reserved for jobs exceeding a total of 1024 parallel processes = tasks. Another queue named big-mem may accomodate tasks with high memory demands by giving access to high-memory nodes (e.g., 512 GB, 1 TB, or more RAM per compute node).
Let’s assume, you have three job queues available, all with identical memory layout:
-
small-jobs: Total task count of up to 511. -
medium-jobs: Total task count 512-1023. -
big-jobs: Total task count of 1024 or more.
When submitting the above computing job, in which queue would it end up? And, if there would be a charge of 1 Cent per core-h, what is the total cost in € (1€ = 100 Cents)?
The total number of tasks results from the product cores-per-node \(\times\) nodes. Total core hours is the task count multiplied by the job’s requested time in hours.
The total number of tasks is cores-per-node \(\times\) nodes = \(64\times 12 = 768\), which would put the
job into themedium-jobsqueue. The HPC center would bill us
for \(64\times 12\times 12 = 9216\)
core hours, hence €92.16.
What are Watt hours?
The unit Wh (Watt-hours) measures energy, so 86,400 Wh is the energy that a 86,400 W (or 86.4 kW, k=kilo) powerful machine consumes in one hour. Back to coffee, brewing one cup needs 50-100 Wh, depending on preparation time and method. So, running your 12-node HPC job for 12 hours is equivalent to brewing between 864 and 1,728 cups of coffee. For those of us who don’t drink coffee, assuming 100% conversion efficiency from our compute job’s heat to mechanical energy, which is unrealistic, we could lift an average African elephant (~6 tons) about 5,285 meters straight up, not quite to the top but in sight of Mount Kilimanjaro’s (5,895 m) summit.
Note that the focus is on extra power, that is, beyond the CPU’s idle state. Attributing our job’s extra power only to CPU usage underestimates its footprint. In practice, the actual delta from idle to full load will vary based on the load posed on other hardware components. Therefore, it is interesting to shed some light onto those other hardware components that start gearing up after hitting that Enter key which submits the above kind of HPC job.
-
CPUs consume power through two main processes:
- Dynamic power consumption: It is caused by the constant switching of transistors and is influenced by the CPU’s clock frequency and voltage.
- Static power consumption: It is caused by small leakage currents even when the CPU is idle. This is a function of the total number of transistors.
Both processes convert electrical energy into heat, which makes CPU cooling so important.
Memory (DRAM) consumes power primarily through its refresh cycles. These are required to counteract the charge leakage in the data-storing capacitors. Periodic refreshing is necessary to maintain data integrity, which is the main reason why DRAM draws power even when idle. Other power consumption factors include the static power drawn by the memory’s circuitry and the active power used during read/write operations.
Network interface cards (NICs) consume power by converting digital data into electrical signals for transmission and reception. Power draw increases with data throughput, physical-media complexity, like fiber optics, and also depends on the specific interconnect technology used.
Storage components: Hard drives (HDDs) require constant energy due to moving mechanical parts, like the disc-spinning motors. SSDs store data electronically via flash memory chips and are thus more power-efficient, especially when idle. However, when performing heavy read/write tasks, SSD power consumption can also be significant, though they complete these tasks faster than HDDs and return to their idle state sooner.
-
Cooling is one of the biggest contributors to total energy use in HPC:
- Idle: Cooling uses ~10–20% of total system power.
- Max load: Cooling can consume ~50–70% of total power (depends on liquid- or air-cooled systems).
Cooling is essential because all electrical circuits generate heat during operation. Under heavy computational loads, insufficiently cooled CPUs and GPUs exceed their safe temperature limits.
These considerations hopefully highlight why there is benefit in identifying potential efficiency bottlenecks before submitting an energy-intense HPC job. If all passengers care about efficient job design, i.e., the total baggage load, more can simultaneously jump onto the HPC plane.
- Using a stopwatch like
timegives you a first tool to log actual versus expected runtimes; it is also useful for carrying out runtime comparisons. - Which hardware piece (CPU, memory/RAM, disk, network, etc.) poses a limiting factor, depends on the nature of a particular application.
- Large-scale computing is power hungry, so we want to use the energy wisely. As shown in the next episodes, you have more power than it may be expected over controlling job efficiency and thus overall energy footprint.
- Computing job efficiency goes beyond individual gain in runtime as shared resources are used more effectively, that is, the ratio \(\frac{useful\;work}{total\;energy\;expended}\sim\frac{number\;of\;users}{total\;energy\;expended}\) improves.
So what’s next?
The following episodes will put a number of these introductory thoughts into concrete action by looking at efficiency aspects around a compute-intense graphical program. While it is not directly an action-loaded video game, it does contain essential pieces thereof, because it uses the technique of ray tracing.
Ray tracing is a technique that simulates how light travels in a 3D scene to create realistic images. It simulates the behaviour of light in terms of optical effects like reflection, refraction, shadows, absorption, etc. The underlying calculations involve real-world physics, which makes them computationally expensive - a perfect HPC case.
Here is a basic run script:
#!/usr/bin/bash
#SBATCH --time=01:00:00
#SBATCH --nodes=1
#SBATCH --tasks-per-node=4
# Put in the same "module load ..." command when building the raytracer program
time mpirun -np 4 raytracer -width=800 -height=800 -spp=128Check thetimeoutput at the end of the job’s output file
(named something like slurm-<NUMBER>.out). You will
notice that user time is by a certain factor larger than
real time.
Why is the user timer larger than the real
time, and what does it mean?
Any guess which number in the mpirunline corresponds
roughly to that factor?
Content from Resource Requirements
Last updated on 2025-12-15 | Edit this page
Overview
Questions
- How many resources should I request initially?
- What scheduler options exist to request resources?
- How do I know if they are used well?
- How large is my HPC cluster?
Objectives
After completing this episode, participants should be able to …
- Identify the size of their jobs in relation to the HPC system.
- Request a good amount of resources from the scheduler.
- Change the parameters to see how the execution time changes.
When you run a program on your local workstation or laptop, you typically don’t plan out the usage of computing resources like memory or core-hours. Your applications simply take as much as they need and if your computer runs out of resources, you can just a few. However, unless you are very rich, you probably don’t have a dedicated HPC cluster just to yourself and instead you have to share one with your colleagues. In such a scenario greedily consuming as many resources as possible is very impolite, so we need to restrain ourselves and carefully allocate just as many resources as needed. These resource constraints are then enforced by the cluster’s scheduling system so that you cannot accidentally use more resources than you think.
Getting a feel for the size of your cluster
To start with your resource planning, it is always a good idea to first get a feeling for the size of the cluster available to you. For example, if your cluster has tens of thousands of CPU cores and you use only 10 of them, you are far away from what would be considered excessive usage of resources. However, if your calculation utilizes GPUs and your cluster has only a handful of them, you should really make sure to use only the minimum amount necessary to get your work done.
Let’s start by getting an overview of the partitions of your cluster:
Here is a (simplified) example output for the command above:
PARTITION NODES CPUS MEMORY GRES TIMELIMIT
normal 223 36 95000+ (null) 1-00:00:00
long 90 36 192000 (null) 7-00:00:00
express 6 36 95000+ (null) 2:00:00
zen4 46 192 763758+ (null) 2-00:00:00
gpuexpress 1 32 240000 gpu:rtx2080:7 12:00:00
gpu4090 8 32 360448 gpu:rtx4090:6 7-00:00:00
gpuh200 4 128 1547843 gpu:h200:8 7-00:00:00In the output, we see the name of each partition, the number of nodes in this partition, the number of CPU cores per node, the amount of memory per node (in Megabytes (or Mebibytes?)), the number of generic resources (typically GPUs) per node and finally the maximum amount of time any job is allowed to take.
Compare the resources available in the different partitions of your local cluster. Can you draw conclusions on what the purpose of each partition is based on the resources it contains?
For our example output above we can make some educated guesses on what the partitions are supposed to be used for:
- The
normalpartition has a (relatively) small amount of memory and limits jobs to at most one day, but has by far the most nodes. This partition is probably designed for small- to medium-sized jobs. Since there are noGRESin this partition, only CPU computations can be performed here. Also, as the number of cores per node is (relatively) small, this partition only allowd multithreading up to 36 threads and requires MPI for a higher degree of parallelism. - The
longpartition has double the memory compared to thenormalpartition, but less than half the number of nodes. It also allows for much longer running jobs. This partition is likely intended for jobs that are too big for thenormalpartition. -
expressis a very small partition with a similar configuration tonormal, but a very short time limit of only 2 hours. The purpose of this partition is likely testing and very short running jobs like software compilation. - Unlike the former partitions,
zen4has a lot more cores and memory per node. The intent of this partition is probably to run jobs using large-scale multithreading. The name of the partitions implies a certain CPU generation (AMD Zen 4), which appears to be newer than the CPU model used in thenormal,longandexpresspartitions (typically core counts increase in newer CPU generations). -
gpuexpressis the first partition that features GPU resources. However, with only a single node and a maximum job duration of 12 hours, this partition seems to be intended again for testing purposes rather than large-scale computations. This also matches the relatively old GPU model. - In contrast,
gpu4090has more nodes and a much longer walltime of seven days and is thus suitable for actual HPC workloads. Given the low number of CPU cores, this partition is intended for GPU workloads only. More details can be gleamed from the GPU model used in this partition (RTX 4090). This GPU type is typically used for Workloads using single-precision floating point calculations. - Finally, the
gpuh200partition combines a large number of very powerful H200 GPUs with a high core count and a very large amount of memory. This partition seems to be intended for the heaviest workloads that can make use of both CPU and GPU resources. The drawback is the low number of nodes in this partition.
To get a point of reference, you can also compare the total number of cores in the entire cluster to the number of CPU cores on the login node or on your local machine.
BASH
lscpu | grep "CPU(s):"
# If lscpu is not available on your machine, you can also use this command
cat /proc/cpuinfo | grep "core id" | wc -lAs you can see, your cluster likely has multiple orders of magnitude more cores in total than the login node or your local machine. To see the amount of memory on the machine you are logged into you can use
Again, the total memory of the cluster is going to be much, much larger than the memory of any individual machine.
All of these cores and all of that memory are shared between you and all the other users of your cluster. To get a feeling for the amount of resources per user, let’s try to get an estimate for how many users there are by counting the number of home directories.
On some clusters, home directories are not placed directly in
/home, but are split up into subdirectories first (e.g., by
first letter of the username like /home/s/someuser). In
this case, you have to use -maxdepth 2 -mindepth 2 to count
the contents of these subdirectories. If your cluster does not use
/home for the users’ home directories, you might have to
use a different path (check dirname "$HOME" for a clue).
Also, this command only gives an upper limit to the number of real
cluster users as there might be home directories for service users as
well.
By dividing the total number of cores / the total memory by the amount of users, you get an estimate of how many resources each user has available in a perfectly fair world.
Does this mean you can never use more than this amount of resources?
Now that you have an idea of how big your cluster is, you can start to make informed decisions on how many resources are reasonable to ask for.
Challenge
sinfo can show a lot more information on the nodes and
partitions of your cluster. Check out the documentation
and experiment with additional output options. Try to find a single
command that will shows for each command the number of allocated and
idle nodes and CPU cores.
BASH
$ sinfo -O Partition,CPUsState,NodeAIOT
PARTITION CPUS(A/I/O/T) NODES(A/I/O/T)
normal* 6336/720/972/8028 196/0/27/223
long 2205/351/684/3240 71/0/19/90
express 44/172/0/216 3/3/0/6
zen4 7532/1108/192/8832 44/1/1/46
gpuexpress 0/32/0/32 0/1/0/1
gpu4090 177/35/44/256 7/0/1/8
gpuh200 90/166/256/512 2/0/2/4Sizing your jobs
The resources required by your jobs primarily depend on the application you want to run and are thus very specific to your particular HPC use case. While it is tempting to just wildly overestimate the resource requirements of your application to make sure it cannot possibly run out, this is not a good strategy. Not only would you have to face the wrath of your cluster administrators (and the other users!) for being inefficient, but you would also be punished by the scheduler itself: In most cluster configurations, your scheduling priority decreases faster if you request more resources and larger jobs often need to wait longer until a suitable slot becomes free. Thus, if you want to get your calculations done faster, you should request just enough resources for your application to work.
Finding this amount of resources is often a matter of trial and error
as many applications do not have precisely predictable resource
requirements. Let’s try this for our snowman renderer. Put the following
in a file named snowman.job:
BASH
#!/bin/bash
#SBATCH --nodes=1
#SBATCH --partition=<put your partition here>
#SBATCH --ntasks=4
#SBATCH --cpus-per-task=1
#SBATCH --mem=1G
#SBATCH --time=00:01:00
#SBATCH --output=snowman-stdout.log
#SBATCH --job-name=snowman
# Always a good idea to purge modules first to start with a clean module environment
module purge
# <put the module load commands for your cluster here>
# Start the raytracer
mpirun -n 4 ./SnowmanRaytracer/build/raytracer -width=1024 -height=1024 -spp=256 -threads=1 -alloc_mode=3 -png=snowman.pngThe #SBATCH directives assign our job the following
resources (line-by-line):
- 1 node…
- … from the partition
<put your partition here> - 4 MPI tasks…
- … each of which uses one CPU core (so 4 cores in total)
- 1 GB of memory
- A timelimit of 1 minute
The last two #SBATCH directives specifiy that we want
the output of our job to be captures in the file
snowman-stdout.log and that the job should appear under the
name snowman.
The --mem directive is somewhat unfortunately named as
it does not define the total amount of memory of your job, but the total
amount of memory per node. Here, this distinction does not
matter as we only use one node, but you should keep in mind that
changing the number of nodes often implies that you need to adapt the
--mem value as well. Alternatively, you can also use the
--mem-per-cpu directive such that the memory allocation
automatically scales with the number of cores. However, even in this
case you need to verify that your memory consumption actually scales
linearly with the number of cores for your application!
To test if our estimate works, you have to submit the job to the scheduler:
This command will also print the ID of the job, so we can observe what is happening with it. Wait a bit and have a look at how your job is doing:
After a while, you will see that the status of your job is given as
TIMEOUT.
You might wonder what the -X flag does in the the
sacct call above. This option instructs Slurm to not output
information on the “job steps” associated with your job. Since we don’t
care about these right now, we set this flag to make the output more
concise.
Check the file snowman-stdout.log as well. Near the
bottom you will see a line like this:
slurmstepd: error: *** JOB 1234567 ON somenode CANCELLED AT 2025-04-01T13:37:00 DUE TO TIME LIMIT ***Evidently our job was aborted because it did not finish within the time limit of one minute that we set above. Let’s try giving our job a time limit of 10 minutes instead.
This time the job should succeed and show a status of “COMPLETED” in
sacct. We can check the resources actually needed by our
job with the help of seff:
The output of seff contains many useful bits of
information for sizing our job. In particular, let’s look at these
lines:
[...]
CPU Utilized: 00:21:34
CPU Efficiency: 98.93% of 00:21:48 core-walltime
Job Wall-clock time: 00:05:27
Memory Utilized: 367.28 MB
Memory Efficiency: 35.87% of 1.00 GBThe exact numbers here depend a lot on the hardware and software of your local cluster.
The Job Wall-clock time is the time our job took. As we
can see, our job takes much longer than one minute to complete which is
why our first attempt with a time limit of one minute has failed.
The CPU Utilized line shows us how much CPU time our job
has used. This is calculated by determining the busy time for each core
and then summing these times for all cores. In an ideal world, the CPU
cores should be busy for the entire time of our job, so the CPU time
should be equal to the time the job took times the number of CPU cores.
The ratio between the real CPU time and the ideal CPU time is shown in
the CPU Efficiency line.
Finally, Memory utilized line shows the peak memory
consumption that your job had at any point during its runtime, while
Memory Efficiency is the ratio between this peak value and
the requested amount of memory for the allocation. As we will see later,
this value has to be taken with a grain of salt.
Starting from the set of parameters that successfully run our
program, we can now try to reduce the amout of requested resources. As
is good scientific practice, we should only vary one parameter at a time
and observe the result. Let’s start by reducing the time limit. There is
often a bit of jitter in the time needed to run a job since not all
nodes are perfectly identical, so you should add a safety margin of 10
to 20 percent (completely arbitrary choice of
numbers here; does everyone agree on the order of magnitude?)
According to the time reported by seff, seven minutes
should therefore be a good time limit. If your cluster is faster, you
might reduce this even further.
As you can see, your job will still complete successfully.
Next, we can optimize our memory allocation. According to SLURM, we used 367.28 MB of memory in our last run, so let’s set the memory limit to 500 MB.
After submitting the job with the lowered memory allocation
everything seems fine for a while. But then, right at the end of the
computation, our job will crash. Checking the job status with
sacct will reveal that the job status is
OUT_OF_MEMORY meaning that our job exceeded its memory
limit and was terminated by the scheduler.
This behavior seems contradictory at first: SLURM reported previously that our job only used around 367 MB of memory at most, which is well below the 500 MB limit we set. The explanation for this discrepancy lies in the fact that SLURM measures the peak memory consumption of jobs by polling, i.e., by periodically sampling how much memory the job currently uses. Unfortunately, if the program has spikes in memory consumption that are small enough to fit between two samples, SLURM will miss them and report an incorrect peak memory value. Spikes in memory usage are quite common, for example if your application uses short-lived subprocesses. Most annoyingly, many programs allocate a large chunk of memory right at the end of the computation to write out the results. In the case of the snowman raytracer, we encode the raw pixel data into a PNG at the end, which means we temporarily keep both the raw image and the PNG data in memory.

SLURM determines memory consuption by polling, i.e.,
periodically checking on the memory consumption of your job. If you job
has a memory allocation profile with short spikes in memory usage, the
value reported by seff can be incorrect. In particular, if
the job gets cancelled due to memory exhaustion, you should not rely on
the value reported by seff as it is likely significantly
too low.
So how big is the peak memory consumption of our process really? Luckily, the Linux kernel keeps track of this for us, if SLURM is configured to use the so-called “cgroups v2” mechanism to enforce resource limits (which many HPC systems are). Let’s use this system to find out how much memory the raytracer actually needs. First, we set the memory limit back to 1 GB, i.e., to a configuration that is known to work.
Next, add these lines at the end of your job script:
BASH
echo -n "Total amount of memory used (in bytes): "
cat /sys/fs/cgroup/$(cat /proc/self/cgroup | awk -F ':' '{print $3}')/memory.peakLet’s break down what these lines do:
- The first line prints out a nice label for our peak memory output.
We use
-nto omit the usual newline thatechoadds at the end of its output. - The second line outputs the contents of a file (
cat). The path of this file starts with/sys/fs/cgroup, which is a location where the Linux kernel exports all the cgroups v2 information as files. - For the next part of the path we need the so-called “cgroup path” of
our job. To find out this path, we can use the
/proc/self/cgroupfile, which contains this path as the third entry of a colon-separated list. Therefore, we read the contents of this file (cat) and extract the third entry of the colon separated list (awk -F ':' '{print $3}'). Since we do this in$(...), Bash will place the output of these commands (i.e., the cgroup path) at this point. - The final part of the path is the information we actually want from
the cgroup. In out case, we are interested in
memory.peak, which contains the peak memory consumption of the cgroup.
When you submit your job and look at the output once it finishes, you will find a line like this:
[...]
Total amount of memory used (in bytes): 579346432
[...]So even though SLURM reported our job to only use 367.28 MB of memory, we actually used nearly 600 MB! With this measurement we can make an informed decision on how to set the memory limit for our job:
Run your job again with this limit to verify that it completes successfully.
So far we have tuned the time and memory limits of our job. Now let
us have a look at the CPU core limit. This limit works slightly
differently than the ones we looked at so far in the sense that your job
is not getting terminated if you try to use more cores than you have
allocated. Instead, the scheduler exploits the fact that multitasking
operating systems can switch out the process a given CPU core is working
on. If you have more active processes in your job than you have CPU
cores (i.e., CPU oversubscription), the operating system will
simply switch processes in and out while trying to ensure that each
process gets an equal amount of CPU time. This happens very fast, so you
can’t see the switching directly, but tools like htop will
show your processes running at less than 100% CPU utilization. Below you
can see a situation of four processes running on three CPU cores, which
results in each process running only 75% of the time.

CPU oversubscription can even be harmful to performance as all the switching between processes by the operating system can cost a non-trivial amount of CPU time itself.
Let’s try reducing the number of cores we allocate by reducing the number of MPI tasks we request in our job script:
Now we have a mismatch between the number of tasks we request and the
number of tasks we use in mpirun. However, MPI catches our
folly and prevents us from accidentally oversubscribing our CPU cores.
In the output file you see the full explanation
There are not enough slots available in the system to satisfy the 4
slots that were requested by the application:
./SnowmanRaytracer/build/raytracer
Either request fewer procs for your application, or make more slots
available for use.
A "slot" is the PRRTE term for an allocatable unit where we can
launch a process. The number of slots available are defined by the
environment in which PRRTE processes are run:
1. Hostfile, via "slots=N" clauses (N defaults to number of
processor cores if not provided)
2. The --host command line parameter, via a ":N" suffix on the
hostname (N defaults to 1 if not provided)
3. Resource manager (e.g., SLURM, PBS/Torque, LSF, etc.)
4. If none of a hostfile, the --host command line parameter, or an
RM is present, PRRTE defaults to the number of processor cores
In all the above cases, if you want PRRTE to default to the number
of hardware threads instead of the number of processor cores, use the
--use-hwthread-cpus option.
Alternatively, you can use the --map-by :OVERSUBSCRIBE option to ignore the
number of available slots when deciding the number of processes to
launch.If we actually want to see oversubscription in action, we need to switch from MPI to multithreading. First, let us try without oversubscribing the CPU cores:
BASH
#SBATCH --ntasks=1
#SBATCH --cpus-per-task=4
# [...]
./SnowmanRaytracer/build/raytracer -width=1024 -height=1024 -spp=256 -threads=4 -alloc_mode=3 -png=snowman.pngThis works and if we look at the output of seff again we
get a baseline for our multithreaded job
[...]
CPU Utilized: 00:21:32
CPU Efficiency: 99.08% of 00:21:44 core-walltime
Job Wall-clock time: 00:05:26
Memory Utilized: 90.85 MB
Memory Efficiency: 12.11% of 750.00 MBChallenge
Compare our measurements for 4 threads here to the measurements we made for doing the computation with 4 MPI tasks earlier. What metrics are similar and which ones are different? Do you have an explanation for this?
We can see that the CPU utilization time and the walltime are virtually identical to the MPI version of our job, while the memory utilization is much lower. The exact reasons for this will be discussed in the following episodes, but here is the gist of it:
- Our job is strongly compute-bound, i.e., the time our job takes is mostly determined by how fast the CPU can do its calculations. This is why it does not matter much for CPU utilization whether we use MPI or threads as long as both can keep the same number of CPU cores busy.
- MPI typically incurs an overhead in CPU usage and memory due to the need to communicate between the tasks (in comparison, threads can just share a block of memory without communication). In our raytracer, this overhead for CPU usage is negligible (hence the same CPU utilization time metrics), but there is a significant memory overhead.
Now let’s see what happens when we oversubscribe our CPU by doubling the number of threads without increasing the number of allocated cores in our job script:
BASH
./SnowmanRaytracer/build/raytracer -width=1024 -height=1024 -spp=256 -threads=8 -alloc_mode=3 -png=snowman.pngChallenge
If you cluster allows direct access to the compute nodes, try logging into the node your job is running on and watch the CPU utilization live using
(Note: Sometimes htop hides threads to make the process
list easier to read. This option can be changed by pressing F2, using
the arrow keys to navigate to the “Hide userlang process threads”,
toggling with the return key and then applying the change with F10.)
Compare the CPU utilization of the raytracter threads
with different total numbers of threads.
In the top right of htop you can also see a metric
called load average. Simplified, this is the number of
processes / threads that are currently either running or could run if a
CPU core was free. Compare the amount of load you generate with your job
depending on the number of threads.
You can see that the CPU utilization of each raytracer
thread goes down as the number of threads increases. This means, each
process is only active for a fraction of the total compute time as the
operating system switches between threads.
For the load metric, you can see that the load increases linearly with the number of threads regardless if they are actually running or waiting for a CPU core. Load is a fairly common metric to be monitored by cluster administrators, so if you cause excessive load by CPU oversubscription you will probably hear from your local admin.
Despite using twice the amount of threads, we barely see any
difference in the output of seff:
CPU Utilized: 00:21:29
CPU Efficiency: 98.85% of 00:21:44 core-walltime
Job Wall-clock time: 00:05:26
Memory Utilized: 93.32 MB
Memory Efficiency: 12.44% of 750.00 MBThis shows that despite having more threads, the CPU cores are not performing more work. Instead, the operating system periodically rotates the threads running on each allocated core, making sure every thread gets a time slice to make progress.
Let’s see what happens when we increase the thread count to extreme levels:
BASH
./SnowmanRaytracer/build/raytracer -width=1024 -height=1024 -spp=256 -threads=1024 -alloc_mode=3 -png=snowman.pngWith this setting, seff yields
CPU Utilized: 00:26:45
CPU Efficiency: 99.07% of 00:27:00 core-walltime
Job Wall-clock time: 00:06:45
Memory Utilized: 113.29 MB
Memory Efficiency: 15.11% of 750.00 MBAs we can see, our job is actually getting slowed down from all the switching between threads. This means, that for our raytracer application CPU oversubscription is either pointless or actively harmful regarding performance.
If CPU oversubscription is so bad, then why do most operating systems default to this behavior?
In this case we have a CPU bound application, i.e., the work done by the CPU is the limiting factor and thus dividing this work into smaller chunks does not help with performance. However, there are also applications bound by other resources. For these applications it makes sense to assign the CPU core elsewhere while the process is waiting, e.g., on a storage medium. Also, in most systems it is desireable to have more programs running than your computer has CPU cores since often only a few of them are active at the same time.
Multi-node jobs
So far, we have only used a single node for our job. The big advantage of MPI as a parallelism scheme is the fact that not all MPI tasks need to run on the same node. Let’s try this with our Snowman raytracer example:
BASH
#!/bin/bash
#SBATCH --nodes=2
#SBATCH --partition=<put your partition here>
#SBATCH --ntasks=4
#SBATCH --cpus-per-task=1
#SBATCH --mem=700M
#SBATCH --time=00:07:00
#SBATCH --output=snowman-stdout.log
#SBATCH --job-name=snowman
# Always a good idea to purge modules first to start with a clean module environment
module purge
# <put the module load commands for your cluster here>
mpirun -- ./SnowmanRaytracer/build/raytracer -width=1024 -height=1024 -spp=256 -threads=1 -alloc_mode=3 -png=snowman.png
echo -n "Total amount of memory used (in bytes): "
cat /sys/fs/cgroup$(cat /proc/self/cgroup | awk -F ':' '{print $3}')/memory.peakThe important change here compared to the MPI jobs before is the
--nodes=2 directive, which instructs Slurm to distribute
the 4 tasks across exactly two nodes.
You can also leave the decision of how many nodes to use up to Slurm by specifying a minimum and a maximum number of nodes, e.g.,
--nodes=1-3would mean that Slurn can assign your job either one, two or three nodes.
Let’s look at the seff report of our job once again:
[...]
Nodes: 2
Cores per node: 2
CPU Utilized: 00:21:32
CPU Efficiency: 98.78% of 00:21:48 core-walltime
Job Wall-clock time: 00:05:27
Memory Utilized: 280.80 MB
Memory Efficiency: 20.06% of 1.37 GBWe can see that Slurm did indeed split up the job such that each of the two nodes is running two tasks. We can also see that the walltime and CPU time of our job are basically the same as before. Considering the fact that communication between nodes is usually much slower than communication within a node, this result is surprising at first. However, we can find an explanation in the way our raytracer works. Most of the compute time is spent on tracing light rays through the scene for each pixel. Since these light rays are independent from one another, there is no need to communicate between the MPI tasks. Only at the very end, when the final image is assembled from the samples calculated by each task, there is some MPI communication happening. The overall communication overhead is therefore vanishingly small.
How well your program scales as you increase the number of nodes depends strongly on the amount of communication in your program.
We can also look at the memory consumption:
[...]
Total amount of memory used (in bytes): 464834560
[...]As we can see, there was indeed less memory consumed on the node running our submit script compared to before (470 MB vs 580 MB). However, our method of measuring peak memory consumption does not tell us about the memory consumption of the second node and we have to use slightly more sophisticated tooling to find out how much memory we actually use.
In the course material is a directory
mpi-cgroups-memory-report that can help us out here, but we
need to compile it first:
Make sure you have a working MPI C Compiler (check with
which mpicc). It is part of the same modules that you need
to run the example raytracer application.
The memory reporting tool works by hooking itself into the
MPI_Finalize function that needs to be called at the very
end of every MPI program. Then, it does basically the same thing as we
did in the script before and checks the memory.peak value
from cgroups v2. To apply the hook to a program, you need to add the
path to the mpi-mem-report.so file we just created to the
environment variable LD_PRELOAD:
BASH
LD_PRELOAD=$(pwd)/mpi-cgroups-memory-report/mpi-mem-report.so mpirun -- ./SnowmanRaytracer/build/raytracer -width=1024 -height=1024 -spp=256 -threads=1 -alloc_mode=3 -png=snowman.pngAfter submitting this job and waiting for it to complete, we can check the output log:
[...]
[MPI Memory Reporting Hook]: Node r05n10 has used 464564224 bytes of memory (peak value)
[MPI Memory Reporting Hook]: Node r07n04 has used 151105536 bytes of memory (peak value)
[...]The memory consumption of the first node matches our previous result, but we can now also see the memory consumption of the second node. Compared to the first node the second node uses much less memory, however in total both nodes use slightly more memory than running all four tasks on a single node (610 MB vs 580 MB). This memory imbalance between the nodes is an interesting observation that we should keep in mind when it comes to estimating how much memory we need per node.
Tips for job submission
To end this lesson, we discuss some tips for choosing resource allocations such that your jobs get scheduled more quickly.
- Many clusters have activated the so-called backfill scheduler option in Slurm. This mechanism tries to squeeze low priority jobs in the gaps between jobs of higher priority (as long as the larger jobs are not delayed by this). In this case, smaller jobs are generally advantageous as they can “skip ahead” in the queue and start early.
- Using
sinfo -t idleyou can specifically search for partitions that have idle nodes. Consider using these partitions for your job if possible as an idle node will typically start your job immediately. - Different partitions might have different billing weights,
i.e., they might use different factors to determine the “cost” of your
calculation, which is subtracted from your compute budget or fairshare
score. You can check these weights using
scontrol show partition <partitionname> | grep TRESBillingWeights. The idea behind different billing weights is to even out the cost of the different resources (i.e., how many hours of memory use correspond to one hour of CPU use) and to ensure that using more expensive hardware carries an appropriate cost for the users. - Typically, it takes longer for a large slot to free up than it takes for several small slots to open. Splitting your job across multiple nodes might not be the most computationally efficient way to run it due to the possible communication overhead, but it can be more efficient in terms of scheduling.
- Slurm produces an estimate on when your job will be started which
you can check with
scontrol show job 35749406 | grep StartTime.
I’m not sure if this is the right section to discuss this…
- Your cluster might seem to have an enormous amout of computing resources, but these resources are a shared good. You should only use as much as you need.
- Resource requests are a promise to the scheduler to not use more
than a specific amount of resources. If you break your promise to the
scheduler and try to use more resources, terrible things will happen.
- Overstepping memory or time allocations will result in your job being terminated.
- Oversubscribing CPU cores will at best do nothing and at worst diminish performance.
- Finding the minimal resource requirements takes a bit of trial and error. Slurm collects a lot of useful metrics to aid you in this.
Content from Scheduler Tools
Last updated on 2025-11-11 | Edit this page
Overview
Questions
- What can the scheduler tell about job performance?
- What’s the meaning of collected metrics?
Objectives
After completing this episode, participants should be able to …
- Explain basic performance metrics.
- Use tools provided by the scheduler to collect basic performance metrics of their jobs.
Scheduler Tools
A scheduler performs important tasks such as accepting and scheduling jobs, monitoring job status, starting user applications, cleaning up jobs that have finished or exceeded their allocated time. The scheduler also keeps a history of jobs that have been run and how they behaved. The information that is collected can be queried by the job owner to learn about how the job utilized the resources it was given.
The seff tool
The seff command can be used to learn about how
efficiently your job has run. The seff command takes the
job identifier as an argument to select which job it displays
information about. That means we need to run a job first to get a job
identifier we can query SLURM about. Then we can ask about the
efficiency of the job.
seff may not be available
seff is an optional SLURM tool for more convenient
access to saact. It does not come standard with every SLURM
installation. Your particular HPC system may or may not provide it.
Check for it’s availability on your login nodes, or consult your cluster
documentation or support staff.
Other third party alternatives, e.g. reportseff, can be installed with default user permissions.
The sbatch command is used to submit a job. It takes a
job script as an argument. The job script contains the resource
requests, such as the amount of time needed for the calculation, the
number of nodes, the number of tasks per node, and so on. It also
contains the commands to execute the calculations.
Using your favorite editor, create the job script
render_snowman.sbatch with the contents below.
#!/usr/bin/bash
#SBATCH --time=01:00:00
#SBATCH --nodes=1
#SBATCH --tasks-per-node=4
# Possibly a "module load ..." command to load required libraries
# Depends on your particular HPC system
mpirun -np 4 raytracer -width=800 -height=800 -spp=128 -alloc_mode=3Next submit the job with sbatch, and see what
seff says about the job with the following commands.
OUTPUT
Job ID: 309489
Cluster: bigiron
User/Group: usr123/grp123
State: COMPLETED (exit code 0)
Nodes: 1
Cores per node: 4
CPU Utilized: 00:07:43
CPU Efficiency: 98.93% of 00:07:48 core-walltime
Job Wall-clock time: 00:01:57
Memory Utilized: 35.75 MB
Memory Efficiency: 0.20% of 17.58 GB (4.39 GB/core)The job script we created asks for 4 CPUs for an hour. After
submitting the job script we need to wait until the job has finished as
seff can only report sensible statistics after the job is
completed. The report from seff shows basic statistics
about the job, such as
- The resources the job was given
- the number of nodes
- the number of cores per node
- the amount of memory per core
- The amount of resources used
-
CPU Utilizedthe aggregate CPU time (the time the job took times the number of CPUs allocated) -
CPU Efficiencythe actual CPU usage as a percentage of the total available CPU capacity -
Job Wall-clock timethe time the job took from start to finish -
Memory Utilizedthe aggregate memory usage -
Memory Efficiencythe actual memory usage as a percentage of the total avaialable memory
-
Looking at the Job Wall-clock time it shows that the job
took just under 2 minutes. Therefore this job took a lot less time than
the one hour we asked for. This can be problematic as the scheduler
looks for time windows when it can fit a job in. Long running jobs
cannot be squeezed in as easily as short running jobs. As a result, jobs
that request a long time to complete typically have to wait longer
before they can be started. Therefore asking for more than 10 times as
much time as the job really needs, simply means that you will have to
wait longer for the job to start. On the other hand you do not want to
ask for too little time. Few things are more annoying than waiting for a
long running calculation to finish, just to see the job being killed
right before the end because it would have needed a couple of minutes
more than you asked for. So the best approach is to ask for more time
than the job needs, but not go overboard here. As the job elapse time
depends on many machine conditions, including congestion in the data
communication, disk access, operating system jitter, and so on, you
might want to ask for a substantial buffer. Nevertheless, asking for
more than twice as much time as job is expected to need, usually doesn’t
make sense.
Another thing is that SLURM by default reserves a certain amount of
memory per core. In this case the actual memory usage is just a fraction
of that amount. We could reduce the memory allocation by explicitly
asking for less by modifying the render_snowman.sbatch job
script.
Challenge
Edit the batch file to reduce the amount of memory requested for the
job. Note that the amount of memory per node can be requested with the
--mem= argument. The amount of memory is specified by a
number followed by a unit. The units can represent kilobtytes (KB),
megabytes (MB), gigabytes (GB). For the calculations we are doing here
100 megabytes per node is more than sufficient. Submit the job, and
inspect the efficiency with seff. What is the memory usage
efficiency you get?
The batch file after adding the memory request becomes.
#!/usr/bin/bash
#SBATCH --time=01:00:00
#SBATCH --nodes=1
#SBATCH --tasks-per-node=4
#SBATCH --mem=100MB
# Possibly a "module load ..." command to load required libraries
# Depends on your particular HPC system
mpirun -np 4 raytracer -width=800 -height=800 -spp=128 -alloc_mode=3Submit this jobscript, as before, with the following command.
OUTPUT
Job ID: 310002
Cluster: bigiron
User/Group: usr123/grp123
State: COMPLETED (exit code 0)
Nodes: 1
Cores per node: 4
CPU Utilized: 00:07:43
CPU Efficiency: 98.09% of 00:07:52 core-walltime
Job Wall-clock time: 00:01:58
Memory Utilized: 50.35 MB
Memory Efficiency: 50.35% of 100.00 MB (100.00 MB/node)The output of seff shows that about 50% of requested
memory was used.
Now we see that a much larger fraction of the allocated memory has been used. Normally you would not worry too much about the memory request. Lately HPC clusters are used more for machine learning work loads which tend to require a lot of memory. Their memory requirements per core might actually be so large that they cannot use all the cores in a node. So there may be spare cores available for jobs that need little memory. In such a scenario tightening the memory allocation up could allow the scheduler to start your job early. How much milage you might get from this depends on the job mix at the HPC site where you run your calculations.
Note that the CPU utilization is reported as almost 100%, but this
just means that the CPU was busy with your job 100% of the time. It does
not mean that this time was well spent. For example, every parallel
program has some serial parts to the code. Typically those parts are
executed redundantly on all cores, which is wasteful but not reflected
in the CPU efficiency. Also, this number does not reflect how well the
capabilities of the CPU are used. If your CPU offers vector
instructions, for example, but your code does not use them then your
code will just run slow. The CPU efficiency will still show that the CPU
was busy 100% of the time even though the program is just running at a
fraction of the speed it could achieve if it fully exploited the
hardware capabilities. It is worth keeping these limitations of
seff in mind.
Good utilization does not imply efficiency
Measuring close to 100% CPU utilization does not say anything about how useful the calculations are. It’s merely stating, that the CPU was mostly busy with calculations, instead of waiting for data or running idle, waiting for other conditions to occur.
Good CPU utilization is only efficient, if it runs only “useful” calculations that contribute with new results towards an intended goal.
The seff command cannot give you any information about
the I/O performance of your job. You have to use other approaches for
that, and sacct may be one of them.
The sacct tool
The sacct command shows data stored in the job
accounting database. You can query the data of any of your previously
run jobs. Just like with seff you will need to provide the
job ID to query the accounting database. Rather than keeping track of
all your jobs yourself you can ask sacct to provide you
with an overview of the jobs you have run.
OUTPUT
JobID JobName Partition Account AllocCPUS State ExitCode
------------ ---------- ---------- ---------- ---------- ---------- --------
309902 render_sn+ STD-s-96h project_a 4 COMPLETED 0:0
309902.batch batch project_a 4 COMPLETED 0:0
309902.exte+ extern project_a 4 COMPLETED 0:0
309903 render_sn+ STD-s-96h project_a 4 COMPLETED 0:0
309903.batch batch project_a 4 COMPLETED 0:0
309903.exte+ extern project_a 4 COMPLETED 0:0
310002 render_sn+ STD-s-96h project_a 4 COMPLETED 0:0
310002.batch batch project_a 4 COMPLETED 0:0
310002.exte+ extern project_a 4 COMPLETED 0:0In the output every job is shown three times here. This is because
sacct lists one line for the primary job entry, followed by
a line for every job step. A job step corresponds to an
mpirun or srun command. The
extern line corresponds to all work that is done outside of
SLURM’s control, for example an ssh command that runs
something somewhere else.
Note that by default sacct only lists the jobs that have
been run today. You can use the --starttime option to list
all jobs that have been run since the given start date. For example, try
running
OUTPUT
JobID JobName Partition Account AllocCPUS State ExitCode
------------ ---------- ---------- ---------- ---------- ---------- --------
308755 snowman.s+ STD-s-96h project_a 16 COMPLETED 0:0
308755.batch batch project_a 16 COMPLETED 0:0
308755.exte+ extern project_a 16 COMPLETED 0:0
308756 snowman.s+ STD-s-96h project_a 4 COMPLETED 0:0
308756.batch batch project_a 4 COMPLETED 0:0
308756.exte+ extern project_a 4 COMPLETED 0:0
309486 interacti+ STD-s-96h project_a 4 FAILED 1:0
309486.exte+ extern project_a 4 COMPLETED 0:0
309486.0 prted project_a 4 COMPLETED 0:0
309489 render_sn+ STD-s-96h project_a 4 COMPLETED 0:0
309489.batch batch project_a 4 COMPLETED 0:0
309489.exte+ extern project_a 4 COMPLETED 0:0
309902 render_sn+ STD-s-96h project_a 4 COMPLETED 0:0
309902.batch batch project_a 4 COMPLETED 0:0
309902.exte+ extern project_a 4 COMPLETED 0:0
309903 render_sn+ STD-s-96h project_a 4 COMPLETED 0:0
309903.batch batch project_a 4 COMPLETED 0:0
309903.exte+ extern project_a 4 COMPLETED 0:0
310002 render_sn+ STD-s-96h project_a 4 COMPLETED 0:0
310002.batch batch project_a 4 COMPLETED 0:0
310002.exte+ extern project_a 4 COMPLETED 0:0You may want to change the date of 2025-09-25 to
something more sensible when you work through this tutorial. Note that
some HPC systems may limit the range of such a request to a maximum of,
for example, 30 days to avoid overloading the slurm database with too
large requests.
With the job ID you can ask sacct for information about
a specific job as in
OUTPUT
JobID JobName Partition Account AllocCPUS State ExitCode
------------ ---------- ---------- ---------- ---------- ---------- --------
310002 render_sn+ STD-s-96h project_a 4 COMPLETED 0:0
310002.batch batch project_a 4 COMPLETED 0:0
310002.exte+ extern project_a 4 COMPLETED 0:0Using sacct with the --jobs flag is just
another way to select which jobs we want more information about. In
itself it does not provide any additional information. To get more
specific data we need to explicitly ask for the information we want. As
SLURM collects a broad range of data about every job it is worth to
evaluate what the most relevant items are.
-
MaxRSS,AveRSS, or the Maximum or Average Resident Size Set (RSS). The RSS is the memory allocated by a program that is actually resident in the main memory of the computer. If the computer gets low on memory then the virtual memory manager can extend the apparently available memory by moving some of the data from memory to disk. This is done entirely transparently to the application, but the data that has been moved to disk is no longer resident in main memory. As a result accessing it will be slower because it needs to retrieved from disk first. Therefore if the RSS is small compared to the total amount of memory the program uses this might affect the performance of the program. -
MaxPages,AvePages, or the Maximum or Average number of Page Faults. These quantities are related to the Resident Size Sets. When the program tries to access data that is not resident in main memory this triggers a page fault. The virtual memory manager responds to a page fault by retrieving the accessed data from disk (and potentially migrating other data to disk to make space). These operations are typically costly. Therefore high numbers of page faults typically correspond to a significant reduction in the program’s performance. For example, the CPU utilization might drop from as high as 98% to as low as 2% due to page faults. For that reason some HPC machines are configured to kill your job if the application generates a high rate of page faults. -
AllocCPUSis the number of CPUs allocated for the job. -
Elapsedis the amount of wall clock time it took to complete the job. I.e. the amount of time that passed between the start and finish of the job. -
MaxDiskRead, the Maximum amount of data read from disk. -
MaxDiskWrite, the Maximum amount of data written to disk. -
ConsumedEnergy, the amount of energy consumed by the job if that information was collected. The data may not be collected on your particular HPC system and is reported as 0. -
AveCPUFreq, the average CPU frequency of all tasks in a job, given in kHz. In general the higher the clock frequency of the processor the faster the calculation runs. The exception is if the application is memory bandwidth limited and the data cannot be moved to processor fast enough to keep it busy. In that case modern hardware might throttle the frequency. This saves energy as the power consumption scales linearly with the clock frequency, but doesn’t slow the calculation down as the processor was having to wait for data anyway.
We can explicitly select the data elements that we are interested in. To see how long the job took to complete run
OUTPUT
Elapsed
----------
00:01:58
00:01:58
00:01:58Challenge
Request information regarding all of the above variables from
sacct, including JobID. Note that the
--format flag takes a comma separated list. Also note that
the result shows that more data is read than written, even though the
program generates and write an image, and reads no data at all. Why
would that be?
To query all of the above variable run
BASH
sacct --jobs=310002 --format=MaxRSS,AveRSS,MaxPages,AvePages,AllocCPUS,Elapsed,MaxDiskRead,MaxDiskWrite,ConsumedEnergy,AveCPUFreqOUTPUT
MaxRSS AveRSS MaxPages AvePages AllocCPUS Elapsed MaxDiskRead MaxDiskWrite ConsumedEnergy AveCPUFreq
---------- ---------- -------- ---------- ---------- ---------- ------------ ------------ -------------- ----------
4 00:01:58 0
51556K 51556K 132 132 4 00:01:58 6.91M 0.72M 0 3M
0 0 0 0 4 00:01:58 0.01M 0.00M 0 3MAlthough the program we have run generates an image and writes that to a file, there is also a none zero amount of data read. The writing part is associated with the image file the program writes. The reading part is not associated with anything that the program does, as it doesn’t read anything from disk. It is instead associated with the fact that the operating system has to read the program itself and it’s dependencies to execute it.
Shortcomings
While seff and sacct provide a lot of
information it is still incomplete. For example, the information is
accumulated for the entire calculation. Variations in the metrics as a
function of time throughout the job are not available. Communication
between different MPI processes is not recorded. The collection of the
energy consumption depends on the hardware and system configuration at
the HPC center and might not be available. We are also often missing
reliable measurements for I/O via the interconnect between nodes and the
parallel file system.
So while we might be able to glean some indications for different types of performance problems, for a proper analysis more detailed information is needed.
Summary
This episode introduced the SLURM tools seff and
sacct to get a high level perspective on a job’s
performance. As these tools just use the statistics that SLURM collected
on a job as it ran, they can always be used without any special
preparation.
Challenge
So far we have considered our initial calculation using 4 cores. To
run the calculation faster we could consider using more cores. Run the
same calculation on 8, 16, and 32 cores as well. Collect and compare the
results from sacct and see how the job performance
changes.
The machine these calculations have been run on has 112 core per node. So we can double the number of cores from 4 until 64 and stay within one node. If we go to two nodes then some of the communication between tasks will have to go across the interconnect. At that point the performance characteristics might change in a discontinuous manner. Hence we try to avoid doing that.
Alternatively you might scale the calculation across multiple nodes, for example 2, 4, 8, 16 nodes. With 112 cores per node you would have to make sure that the calculation is large enough for such a large number of cores to make sense.
Create running_snowmen.sh with
#!/usr/bin/bash
for nn in 4 8 16 32; do
id=`sbatch --parsable --time=00:12:00 --nodes=1 --tasks-per-node=$nn --ntasks-per-core=1 render_snowman.sh`
echo "ntasks $nn jobid $id"
doneCreate render_snowman.sh with
#!/usr/bin/bash
# Possibly a "module load ..." command to load required libraries
# Depends on your particular HPC system
export START=`pwd`
# Create a sub-directory for this job if it doesn't exist already
mkdir -p $START/test.$SLURM_NTASKS
cd $START/test.$SLURM_NTASKS
# The -spp flag ensures we have enough samples per ray such that the job
# on 32 cores takes longer than 30s. Slurm by default is configured such
# that job data is collected every 30s. If the job finishes in less than
# that Slurm might fail to collect some of the data about the job.
mpirun -np $SLURM_NTASKS raytracer -width=800 -height=800 -spp 1024 -threads=1 -alloc_mode=3 -png=rendered_snowman.pngNext we submit this whole set of calculations
producing
OUTPUT
ntasks 4 jobid 349291
ntasks 8 jobid 349292
ntasks 16 jobid 349293
ntasks 32 jobid 349294After the jobs are completed we can run
BASH
sacct --jobs=349291,349292,349293,349294 \
--format=MaxRSS,AveRSS,MaxPages,AvePages,AllocCPUS,Elapsed,MaxDiskRead,MaxDiskWrite,ConsumedEnergy,AveCPUFreqto produce
OUTPUT
MaxRSS AveRSS MaxPages AvePages AllocCPUS Elapsed MaxDiskRead MaxDiskWrite ConsumedEnergy AveCPUFreq
---------- ---------- -------- ---------- ---------- ---------- ------------ ------------ -------------- ----------
4 00:09:35 0
142676K 142676K 1 1 4 00:09:35 7.75M 0.72M 0 743K
0 0 0 0 4 00:09:35 0.01M 0.00M 0 2.61M
8 00:05:01 0
289024K 289024K 0 0 8 00:05:01 10.15M 1.45M 0 960K
0 0 0 0 8 00:05:02 0.01M 0.00M 0 2.42M
16 00:02:21 0
563972K 563972K 93 93 16 00:02:21 15.00M 2.94M 0 1.03M
0 0 0 0 16 00:02:21 0.01M 0.00M 0 2.99M
32 00:01:14 0
1082540K 1082540K 260 260 32 00:01:14 24.83M 6.07M 0 1.08M
0 0 0 0 32 00:01:14 0.01M 0.00M 0 3MNote that the elapse time goes down as the number of cores increases,
which is reasonable as more cores normally can get the job done quicker.
The amount of data read also increases as every MPI rank has to read the
executable and all associated shared libraries. The volume of data
written is harder to understand. Every run produces an image file
rendered_snowman.png that is about 100KB in size. This file
is written just by the root MPI rank. This cannot explain the increase
in data written with increasing numbers of cores. The increasing number
of page faults with increasing numbers of cores suggests that paging
memory to disk is responsible for the majority of data written.
- Schedulers provide tools for a high level view on our jobs,
e.g.
sacctandseff - Important basic performance metrics we can gather this way are:
-
CPU Utilization, often as fraction of
time where CPU was active/elapsed time of the job - Memory utilization, often measured as Resident Set Size (RSS) and number of Pages
-
CPU Utilization, often as fraction of
-
sacctcan also provide metrics about disk I/O and energy consumption - Metrics through
sacctare accumulated for the whole job runtime and may be too broad for more specific insight
Content from Scaling Study
Last updated on 2025-12-12 | Edit this page
Overview
Questions
- How many resources should be requested for a given job?
- How does our application behave at different scales?
Objectives
After completing this episode, participants should be able to …
- Perform a scaling study for a given application.
- Notice different perspectives on scaling parameters.
- Identify good working points for the job configuration.
The deadline is approaching way too fast and we may not finish our project in time. Maybe requesting more resources from our clusters scheduler does the trick? How could we know if it helps and by how much?
What is Scaling?
The execution time of parallel applications changes with the number of parallel processes or threads. In a scaling study we measure how much the execution time changes by scanning a reasonable range of number of processes. In a common phrasing, this approach answers how the execution time scales with the number of parallel processors.
Starting from the job script render_snowman.sbatch:
BASH
#!/usr/bin/bash
#SBATCH --time=01:00:00
#SBATCH --nodes=1
#SBATCH --mem-per-cpu=200MB
# The `module load` command you had to load for building the raytracer
module load 2025 GCC/13.2.0 OpenMPI/4.1.6 buildenv/default Boost/1.83.0 CMake/3.27.6 libpng/1.6.40
time mpirun -- ./raytracer -width=800 -height=800 -spp=128 -png "$(date +%Y-%m-%d_%H%M%S).png"we can manually run such a scaling study by submitting multiple jobs.
In OpenMPI versions 4 and 5 the number of Slurm tasks is automatically
picked up, so we do not set -n or -np of
mpirun. We use -- to separate the arguments of
mpirun – none in this case – from the MPI application
raytracer and its arguments. Otherwise you may experience
errors in some versions of OpenMPI 5, where mpirun
misinterprets the arguments of raytracer as its own.
Scaling other resources with number of CPU cores
When scaling the resources outside of the job script, e.g. with
sbatch --ntasks=X ..., as done above, we make sure to scale
other resource requirements with the number of parallel processors. In
this case, --mem-per-cpu=200MB is necessary, since
--mem results in a fixed memory limit, independent of the
number of processes.
For example, if each MPI process needs \(100\,\)MB, requesting \(2\,\)GB would only be enough for up to 20 MPI processes.
Forgetting a limit like this is a common pitfall in this situation.
Let’s start some measurements with \(1\), \(2\), \(4\), and \(8\) tasks:
OUTPUT
$ sbatch --ntasks 1 render_snowman.sbatch
Submitted batch job 16142767
$ sbatch --ntasks 2 render_snowman.sbatch
Submitted batch job 16142768
$ sbatch --ntasks 4 render_snowman.sbatch
Submitted batch job 16142769
$ sbatch --ntasks 8 render_snowman.sbatch
Submitted batch job 16142770Now we have to wait until all four jobs are finished.
Regular update of squeue
You can use squeue --me -i 30 to get an update of all of
your jobs every 30 seconds.
If you don’t need a more regular update, it is good practice to keep the interval on the order of 30s to a couple of minutes, just to be nice to Slurms server resources.
Once the jobs are finished, we can use grep to get the
wall clock time of all four jobs:
OUTPUT
slurm-16142767.out:real 2m7.218s
slurm-16142768.out:real 1m7.443s
slurm-16142769.out:real 0m32.584s
slurm-16142770.out:real 0m17.480sThe real-time is decreasing significantly each time we double the number of Slurm tasks. From this, we feel that doubling the number of CPU cores really is a winning strategy!
Exercise: Continue scaling study to larger values
Run the same scaling study and continue it for even larger number of
--ntasks, e.g. 16, 32, 64, 128. So far, we have been using
--nodes=1 to stay on a single node. At which point are your
MPI processes distributed across more than one node? Use Slurm command
line tools to find out the how many CPU cores (MPI processes) are
available on a single node. You may have to increase the number of nodes
with --nodes, if you want to go beyond that limit.
Gather your real time results and place them in a
.csv file. Here is an example for our previous
measurements:
ntasks,time
1,127.218
2,67.443
4,32.584
8,17.480
...How much does each doubling of the CPU resources help with running the parallel raytracer?
You can use sinfo to find out the node names of your
particular Slurm partition. Then use scontrol to show all
details about a single node from that partition. It will show you the
number of CPU (cores) available on that node.
ntasks,time
1,127.218
2,67.443
4,32.584
8,17.480
16,10.251
32,7.257
64,8.044
128,8.575Using grep "real" slurm-*.out, we can see the execution
time is halved in the beginning, with each doubling of the CPU cores.
However, somewhere between \(8\) and
\(16\) cores, we start to see less and
less improvement.
Adding more resources does not help indefinitely. At some point the overhead of managing the calculation in separate tasks outweighs the benefit of parallel calculation. There is too little to do in each tasks and the overhead starts to dominate.
At some point adding more CPU cores does not help us anymore.
Adding more CPU cores can actively slow down the calculation after a certain point. The optimal point is different for each application and each configuration. It depends on the ratio between calculations, communications and various management overheads in the whole process of running everything.
Overheads and Reliable Measurements
Many overheads and when they show also depend on the underlying hardware. So the sweet spot may very well be different for different clusters, even if the application and configuration stays the same!
Another common issue lies within our measurements themself. We
perform a single time measurement on a worker node that is possibly
shared with other jobs at the same time. What if another user runs an
application that hogs shared resources like the local disk or network
interface card? In this case our measurements become somewhat
non-deterministic. Running the same measurement twice may result in
significantly different values. If you need reliable results, e.g. for a
publication, requesting exclusive access to Slurms resources through the
sbatch flag --exclusive is the best approach.
As a drawback, this typically results in longer waiting times, since
whole nodes have to be reserved for the measurement jobs, even if not
all resources are used.
Even on exclusive resources, the measurements cannot be 100% reliable. For example, the scheduling behavior of the Linux kernel, or access to remote resources like the parallel file system or data from the web, are still affecting your measurements in unpredictable ways. Therefore, the best results are achieved by taking the mean and standard deviations of repeated measurements for the same configuration. The measured minimum also has strong informative value, since it proofs the best observed behavior.
Keep in mind, --exclusive will always request all
resources of a given node, even if only few cores are used. In these
cases, tools like seff show worse resource utilization
results, since measurements are done with respect to all booked
resources.
Scaling studies can be done with respect to different application and job parameters. For example, what is the execution time when we change the workload, e.g. a larger number of pixels, samples per pixels, or a more complex scene? How much does a communication overhead change, if we change the number of involved nodes while keeping the workload and number of tasks fixed, i.e. changing the network communication surface? Scaling studies like these can help identify pressure points that affect the applications performance.
Scaling studies typically occur in a preparation phase where the application is evaluated with a representative example workload. Once a good configuration is found, we know the application is running close to an optimal performance and larger number of calculations can start, often called the production phase.
In a similar vein, scaling studies can be a formal requirement for compute time applications on larger HPC systems. On these systems and for larger calculation campaigns it is more crucial to run efficient calculations, since the resources are typically more contested and the potential energy- and carbon footprint becomes much larger.
Speedup, Efficiency, and Strong Scaling
To quantitatively and empirically study the scaling behavior of a given application, it is common to look at the speedup and efficiency with respect to adding more parallel processors.
Speedup is a metric to compare the execution times with different amounts of resources. It answers the question
How much faster is the application with \(N\) parallel processes/threads, compared to the serial execution with \(1\) process/thread)?
It is defined by the comparison of wall times \(T(N)\) of the application with \(N\) parallel processes: \[S(N) = \frac{T(1)}{T(N)}\] Here, \(T(1)\) is the wall time for a sequential execution, and \(T(N)\) is the execution with \(N\) parallel processes. For \(2\) processes, we observe a speedup of \(S(2) = \frac{127.218}{67.443} \approx 1.89\)
Efficiency in this context is defined as \[\eta(N) = \frac{S(N)}{N}\] with speedup \(S(N)\) and describes by how much additional parallel processes, \(N\), deviate from the theoretical linear optimum.
Exercise: Calculate Speedup and Efficency
Extend the .csv file of your measurements from above
with a speedup and efficiency column. It may
look like this:
ntasks,time,speedup,efficiency
1,127.218,1.00,1.00
2,67.443,1.89,0.94
4,32.584,3.90,0.98
8,17.480,7.28,0.91
...You may want to use any data visualization tool, e.g. python or spreadsheets, to visualize the data.
What number of processes may be a good working point for the raytracer with \(800 \times 800\) pixel and \(128\) samples per pixel?
For all of our measurements, the speedup and efficiencies are
ntasks,time,speedup,efficiency
1,127.218,1.00,1.00
2,67.443,1.89,0.94
4,32.584,3.90,0.98
8,17.480,7.28,0.91
16,10.251,12.41,0.78
32,7.257,17.53,0.55
64,8.044,15.82,0.25
128,8.575,14.84,0.12Plotting the speedup and efficiency helps with identifying a good working point:
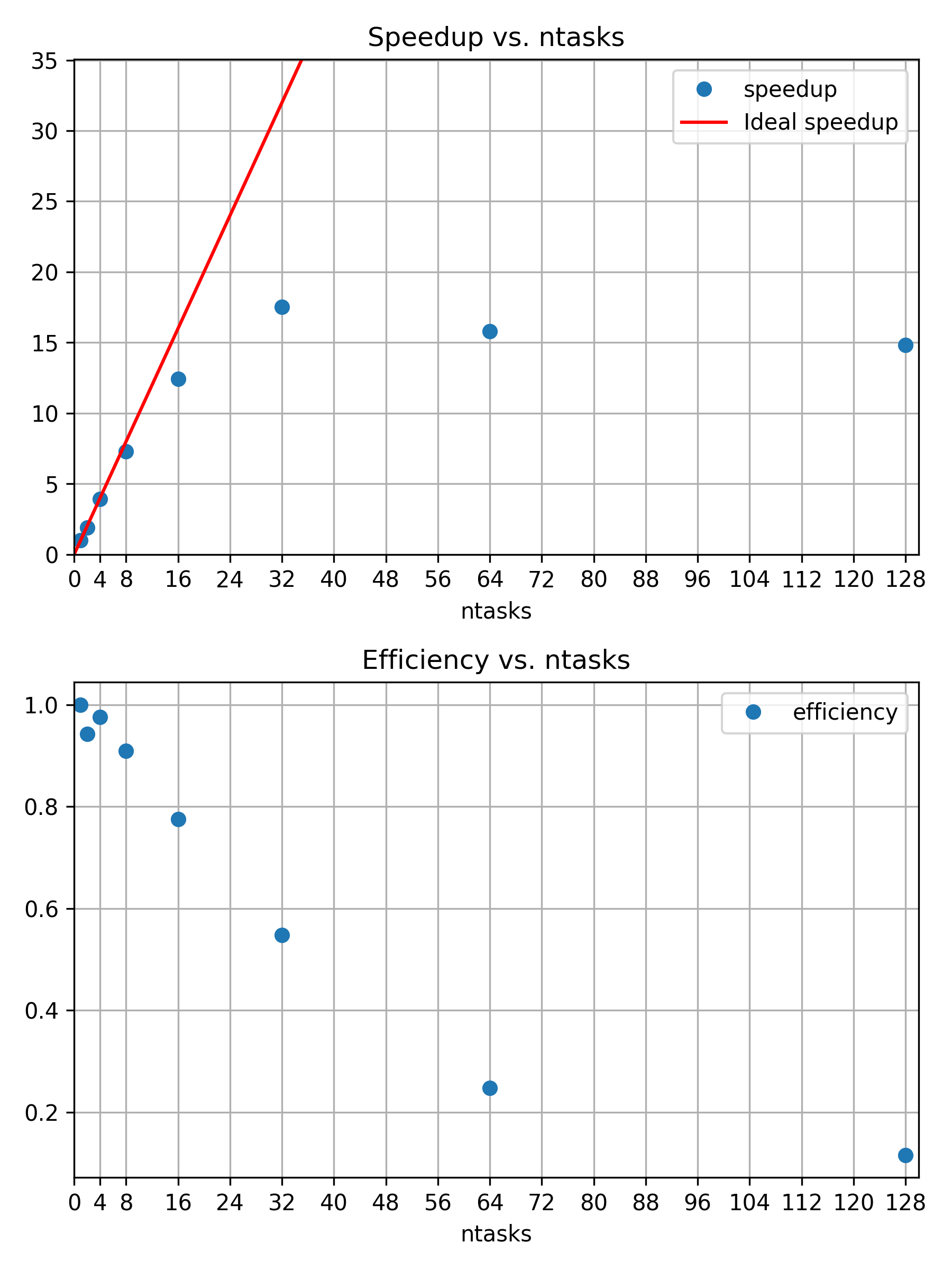
The 16th processor is still close to 80% efficient. The corresponding speedup is less than the theoretical optimum, which is visualized by a red line of slope \(1\).
There is no exact optimum and the best working point is open for discussion. However, it would be difficult to justify additional cores, if their contribution to speedup is only 50% efficient or even less.
If you have experience with python, you can use our python script to create the
same plots as above, but for your own data. It depends on
numpy, pandas, and matplotlib, so
make sure to prepare a corresponding python environment.
The script expects your .csv files to be called
strong.csv and weak.csv, and be placed in the
same directory.
So far, we kept the workload size fixed to \(800 \times 800\) pixels and \(128\) samples per pixel for the same scene with three snowman. The diminishing returns for adding more and more parallel processors leads to a famous observation. The speedup of a program through parallelization is limited by the execution time of the serial fraction that is not parallelizable. No application is 100% parallelizable, so adding an arbitrary amount of parallel processors can only affect the parallelizable section. In the best case, the execution time gets reduced to the serial fraction of the application.
An application is said to scale strongly, if adding more cores significantly reduces the execution time.
Amdahls Law1
The speedup of a program through parallelization is limited by the execution time of the serial fraction that is not parallelizable. For a given execution time \(T(N) = s + \frac{p}{N}\), with \(s\) the time for the serial fraction, and \(p\), the time for parallel fraction, speedup \(S\) is defined as \[S(N) = \frac{s+p}{s+\frac{p}{N}} = \frac{1}{s + \frac{p}{N}} \Rightarrow \lim_{N\rightarrow \infty} S(N) = \frac{1}{s}\]
Discussion: When should we stop adding CPU cores?
Discuss your previous results and decide on a good working point. How many cores are still usefully reducing the execution time.
What other factors could affect your decision, e.g. available hardware and corresponding waiting times.
If scaling is limited, why are there larger HPC systems? Weak scaling.
For a fixed problem size, we observed that adding more parallel processors can only help up to a certain point. But what if the project benefits from increasing the workload size? Does a higher resolution, more accuracy, or more statistics, etc., improve our insights and results? If that is the case, the perspective on the issue changes and adding more parallel processors can become more feasible as well. For our raytracer example, increasing the workload corresponds to more pixels, more samples per pixel, and/or a more complex scene.
Weak scaling refers to the scaling behavior of an application for a fixed workload per parallel processing unit, e.g. increasing the number of pixels by the same amount as the number of parallel processors \(N\).
Gustafsons Law2
A program scales on \(N\) parallel processors, if the problem size also scales with the number of processors. The speedup \(S\) becomes \[\text{S(N)} = \frac{s+pN}{s+p} = s+pN = N+s(1-N)\] with \(N\) processors, \(s\) the time for the serial fraction, and \(p\), the time for parallel fraction:
To scale the workload of the snowman raytracer, we can increase the number of calculated pixels with the same factor with which we increase the number of parallel processors. For one processor we have \(800 \times 800 = 640000\) pixel. That means for two processors we need a height and a width of \(\sqrt{2 \times 640000} = 1131.371 \approx 1131\). And similarly increasing the number of pixels for \(--ntasks=4\) and so on.
The job script could look like this:
BASH
#!/usr/bin/bash
#SBATCH --time=01:00:00
#SBATCH --nodes=1
#SBATCH --mem-per-cpu=3800MB
module load 2025 GCC/13.2.0 OpenMPI/4.1.6 buildenv/default Boost/1.83.0 CMake/3.27.6 libpng/1.6.40
# Create associative array
declare -A pixel
pixel[1]="800"
pixel[2]="1131"
pixel[4]="1600"
pixel[8]="2263"
pixel[16]="3200"
pixel[32]="4526"
pixel[64]="6400"
time mpirun -- ./build/raytracer -width=${pixel[${SLURM_NTASKS}]} -height=${pixel[${SLURM_NTASKS}]} -spp=128 -threads=1 -png "$(date +%Y-%m-%d_%H%M%S).png"To scale the workload of the snowman raytracer, we can multiply the
number of parallel MPI processes, ${SLURM_NTASKS}, with the
samples per pixel (starting from -spp=128). For a single
process, the whole \(800 \times 800\)
pixel picture is calculated in a single MPI process with 128 per pixel.
Running with two MPI processes, both have to calculate half the number
of pixels, but twice the amount of samples per pixel.
BASH
#!/usr/bin/bash
#SBATCH --time=01:00:00
#SBATCH --nodes=1
#SBATCH --mem-per-cpu=500MB
module load 2025 GCC/13.2.0 OpenMPI/4.1.6 buildenv/default Boost/1.83.0 CMake/3.27.6 libpng/1.6.40
SPP="$[${SLURM_NTASKS}*128]"
time mpirun -- ./build/raytracer -width=800 -height=800 -spp=${SPP} -threads=1 -png "$(date +%Y-%m-%d_%H%M%S).png"

In direct comparison, and zooming in really close, you can see more noise in the first image, e.g. in the shadows. One could argue that we passed the point of diminishing returns, though. Is a \(64\times\) increase in computational cost worth the observed quality improvement? For the samples per pixel, we seem to not benefit much from weak scaling. Larger resolutions, by increasing the number of pixels, is the more useful dimension to increase in this case.
Increasing the resolution may be worth the effort, if we have a use for a larger, more detailed picture. In practice, there is a cutoff, beyond which no reasonable improvement is to be expected. This is a question about accuracy, error margins, and overall quality, which can only be answered in the specific context of each research project. If there is no real improvement by increasing the workload, running a weakly scaling application is really just wasting valuable computational time and energy.
If we increase the workload at the rate as our number of parallel processes (\(N\)) our speedup is defined as \[S_{\text{weak}}(N) = \frac{T(1)}{T(N)} \times N\] since we do \(N\) times more work with \(N\) processors, compared to our reference \(T(N=1)\). Efficiency is still defined as \[\eta_{\text{weak}}(N) = \frac{S_{\text{weak}}(N)}{N} = \frac{T(1)}{T(N)}\]
Exercise: Weak scaling
Repeat the previous scaling study and increase the number of pixels accordingly to study the raytracers weak scaling behavior.
- Run with 1, 2, 4, 8, 16, 32, 64 MPI processes on single node
- Take
timemeasurements and consider running with--exclusiveto ensure more reliable results. - Create a
.csvfile and run the plotting script
ntasks,pixel,time,speedup,efficiency
1,800,123.162
2,1131,122.562
4,1600,124.522
8,2263,125.606
...How well does the application scale with an increasing workload size?
Do you see a qualitative difference in the resulting .png
files and is the increased sample-per-pixel size worth the computational
costs?
ntasks,pixel,time,speedup,efficiency
1,800,123.162
2,1131,122.562
4,1600,124.522
8,2263,125.606
16,3200,125.803
32,4526,130.137
64,6400,138.636The scaling behavior is reaching an asymptotic limit, where each additional processor is contributing with the same efficiency to the increased workload.
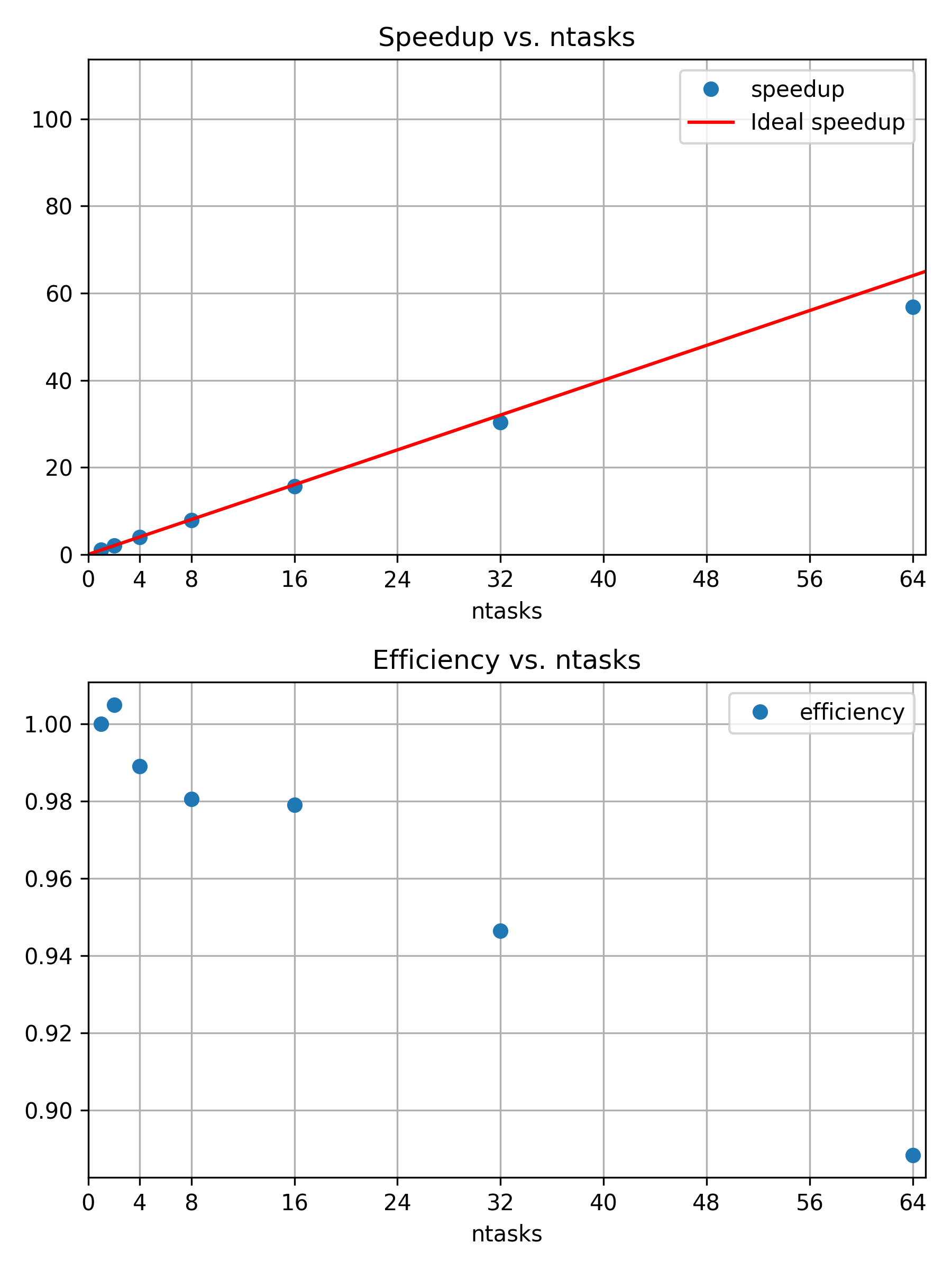
Weakly scaling jobs can make efficient use of a huge amount of resources.
The most important question is, if an increased workload is producing useful results. Here, we have the rendered picture of three snowmen in 800x800 with 128 samples per pixel and three snowmen in 6400x6400 with 128 samples per pixel. The second image has a much higher resolution. However, going way beyond \(6400 \times 6400\) pixels is probably not very meaningful, unless you are trying to print the worlds largest ad boards or similar.
{kind=link}
{kind=link}
Summary
In this episode, we have seen that we can study the scaling behavior of our application with respect to different metrics, while varying its configuration. Most commonly, we study the execution time of an application with an increasing number of parallel processors. In such a scaling study, we collect comparable walltime measurements for an increasing number of Slurm tasks of a parallelizable and representative job. If a good working point is found, larger scale “production” jobs can be submitted to the HPC system.
If the application has good strong scaling behavior, adding more cores leads to an effective improvement in execution time. We observe diminishing returns of adding more cores to a fixed-size problem, so there is a (subjective) optimal number of parallel processors for a given application configuration. (Amdahls Law)
If increasing the workload size leads to better results, maybe because of improved accuracy and quality, we can study the weak scaling behavior and increase the workload size by the same factor of increasing parallel processors.
A good working point depends on the availability of resources, specifics of the underlying hardware, the particular application, and a particular configuration for the application. For that reason, scaling studies are a common requirement for formal compute time applications to prove an efficient execution of a given application.
We can study the impact of any parameter on metrics like, for example, walltime, CPU utilization, FLOPS, memory utilization, communication, output size on disk, and so on.
If you find yourself repeating similar measurements over and over again, you may be interested in an automation approach. This can be done by creating reproducible HPC workflows using JUBE, among other things.
Up to now, we were still working with basic metrics like the wall-clock time. In the next episode, we start with more in-depth measurements of many other aspects of our job and application.
- Jobs behave differently with increasing parallel resources and fixed or scaling workloads
- Scaling studies can help to quantitatively grasp this changing behavior
- Good working points are defined by configurations where more cores still provide sufficient speedup or improve quality through increasing workloads
- Amdahl’s law: speedup is limited by the serial fraction of a program
- Gustafson’s law: more resources for parallel processing still help, if larger workloads can meaningfully contribute to project results
G. M. Amdahl, ‘Validity of the single processor approach to achieving large scale computing capabilities’, in Proceedings of the April 18-20, 1967, spring joint computer conference, in AFIPS ’67 (Spring). New York, NY, USA: Association for Computing Machinery, Apr. 1967, pp. 483–485. doi: 10.1145/1465482.1465560.↩︎
J. L. Gustafson, ‘Reevaluating Amdahl’s law’, Commun. ACM, vol. 31, no. 5, pp. 532–533, May 1988, doi: 10.1145/42411.42415.↩︎
Content from Performance Overview
Last updated on 2025-11-11 | Edit this page
Overview
Questions
- Is it enough to look at a jobs walltime?
- What steps can I take to evaluate a jobs performance?
- What popular types of reports exist?
Objectives
After completing this episode, participants should be able to …
- Explain different approaches to performance measurements.
- Understand common terms and concepts in performance analyses.
- Create a performance report through a third-party tool.
- Describe what a performance report is meant for (establish baseline, documentation of issues and improvements through optimization, publication of results, finding the next thread to pull in a quest for optimization)
- Measure the performance of central components of underlying hardware (CPU, Memory, I/O, …) (split episode?)
- Identify which general areas of computer hardware may affect performance.
Workflow
- Previously checked scaling behavior by looking at walltime
- what if we would count other things while our job is running? Could
be
- CPU utilization
- FLOPS
- Memory uitilization
- …
- Two possible ways to look at this data with respect to time:
- tracing: over time
- sampling: accumulated results at the end
- Third-party tools to measure these things - you can use them with your jobs
Here you can choose between three alternative perspectives on our job:
- ClusterCockpit: A job monitoring service available on many of our clusters. Needs to be centrally maintained by your HPC administration team.
- Linaro Forge Performance Reports: A commercial application providing a single page performance overview of your job. Your cluster may have licenses available.
- TBD: A free, open source tool/set of tools, to get a general performance overview of your job.
Performance counters and permissions, may require
--exclusive, depends on system! Look at documentation /
talk to your administrators / support.
cap_perfmon,cap_sys_ptrace,cap_syslog=ep
kernel.perf_event_paranoidLive coding:
- Set up the main tool. How do I access it? How can I use it with my job?
- Run snowman with 8 cores
General report
How Does Performance Relate to Hardware?
(Following this structure throughout the course, trying to understand the performance in these terms)
Broad dimensions of performance:
- CPU (Front- and Backend, FLOPS)
- Frontend: decoding instructions, branch prediction, pipeline
- Backend: getting data from memory, cache hierarchy & alignment
- Raw calculations
- Vectorization
- Out-of-order execution
- Accelerators (e.g. GPUs)
- More calculations
- Offloading
- Memory & communication models
- Memory (data hierarchy)
- Working memory, reading data from/to disk
- Bandwidth of data
- I/O (broader data hierarchy: disk, network)
- Stored data
- Local disk (caching)
- Parallel fs (cluster-wide)
- MPI-Communiction
- Parallel timeline (synchronization, etc.)
- Application logic
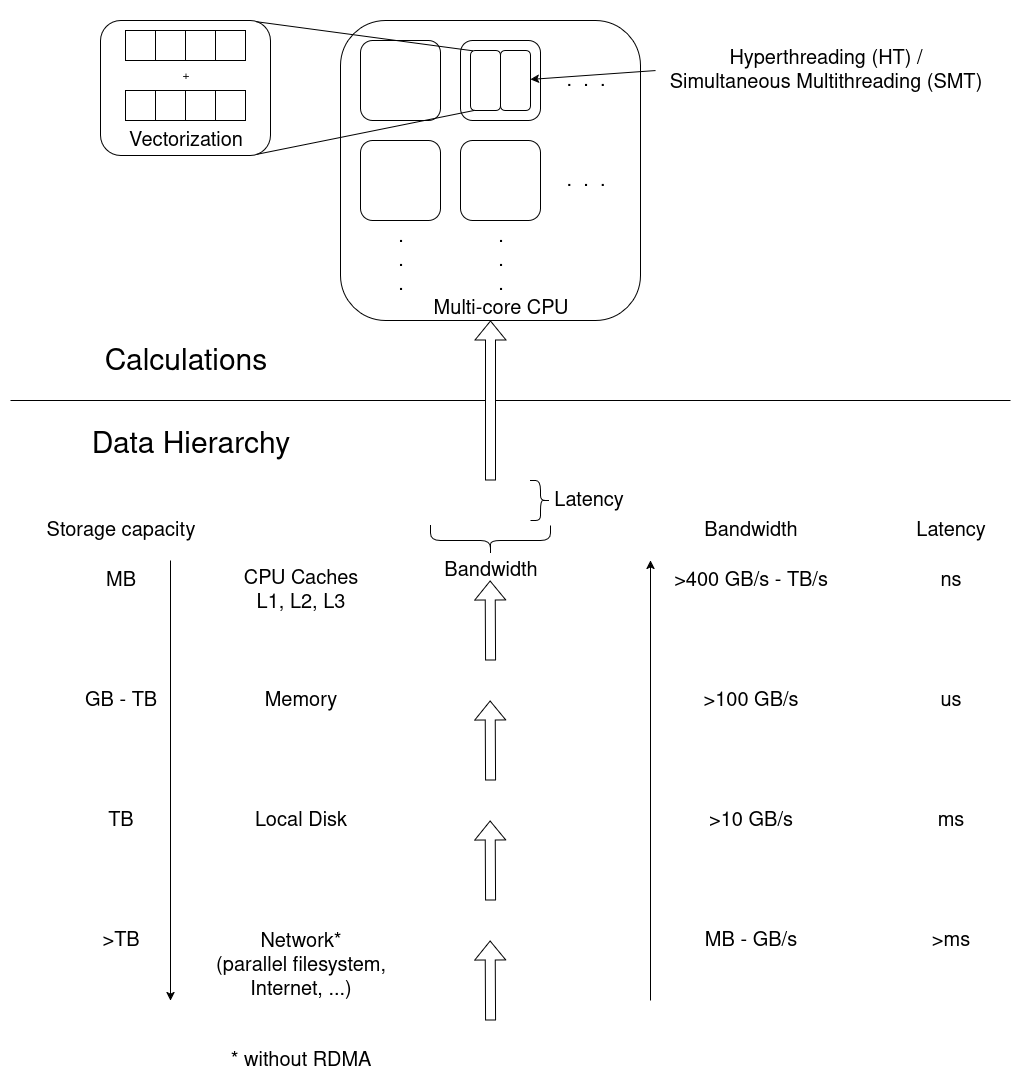
Exercise: Match application behavior to hardware
Which part of the computer hardware may become an issue for the following application patterns:
- Calculating matrix multiplications
- Reading data from processes on other computers
- Calling many different functions from many equally likely if/else branches
- Writing very large files (TB)
- Comparing many different strings if they match
- Constructing a large simulation model
- Reading thousands of small files for each iteration
Maybe not the best questions, also missing something for accelerators.
- CPU (FLOPS) and/or Parallel timeline
- I/O (network)
- CPU (Front-End)
- I/O (disk)
- (?) CPU-Backend, getting strings through the cache?
- Memory (size)
- I/O (disk)
Summary
- General reports show direction in which to continue
- Specialized tools may be necessary to move on
Leading question: Connection to hardware is quite deep, why does it matter? -> Drill deeper, e.g. on NUMA & pinning
- First things first, second things second, …
- Profiling, tracing
- Sampling, summation
- Different HPC centers may provide different approaches to this workflow
- Performance reports offer more insight into the job and application behavior
Content from Pinning
Last updated on 2025-10-31 | Edit this page
Overview
Questions
- What is “pinning” of job resources?
- How can pinning improve the performance?
- How can I see, if pinning resources would help?
- What requirement hints can I give to the scheduler?
Objectives
After completing this episode, participants should be able to …
- Define the concept of “pinning” and how it can affect job performance.
- Name Slurms options for memory- and cpu- binding.
- Use hints to tell Slurm how to optimize their job allocation.
Binding / pinning:
--mem-bind=[{quiet|verbose},]<type>-m, --distribution={*|block|cyclic|arbitrary|plane=<size>}[:{*|block|cyclic|fcyclic}[:{*|block|cyclic|fcyclic}]][,{Pack|NoPack}]-
--hint=: Hints for CPU- (compute_bound) and memory-bound (memory_bound), but alsomultithread,nomultithread -
--cpu-bind=[{quiet|verbose},]<type>(srun) - Mapping of application <-> job resources
Motivation
Exercise
Case 1: 1 thread per rank
mpirun -n 8 ./raytracer -width=512 -height=512 -spp=128 -threads=1 -alloc_mode=3 -png=snowman.png
Case 2: 2 thread per rank
mpirun -n 8 ./raytracer -width=512 -height=512 -spp=128 -threads=2 -alloc_mode=3 -png=snowman.png
Questions: - Do you notice any difference in runtime between the two cases? - Is the increase in threads providing a speedup as expected?
- Observation: The computation times are almost the same.
- Expected behavior: Increasing threads should ideally reduce runtime.
- Hypothesis: Additional threads do not contribute.
How to investigate?
You can verify the actual core usage in two ways: 1. Use
--report-bindings with mpirun 2. Use
htopcommand on the compute node
Exercise
Follow any one of the option above and run for 2 threads per rank
mpirun -n 8 ./raytracer -width=512 -height=512 -spp=128 -threads=2 -alloc_mode=3 -png=snowman.png
Questions: - Did you find any justification for the hypothesis we made?
Only 8 cores are active instead of 16
Explanation:
- Eventhough we requested 2 threads per MPI rank, both threads are pinned to the same core.
- The second thread waits for the first thread to finish, so no actual thread-level parallelization is achieved.
How to achieve?
Exercise: Understanding Process and Thread Binding
Pinning (or binding) means locking a process or thread to a specific hardware resource such as a CPU core, socket, or NUMA region. Without pinning, the operating system may move tasks between cores, which can reduce cache reuse and increase memory latency, directly diminishes performance.
In this exercise we will explore how MPI process and thread binding works. We will try binding to core, socket, and numa, and observe timings and bindings.
Exercise
Case 1: --bind-to numa
mpirun -n 8 --bind-to numa ./raytracer -width=512 -height=512 -spp=128 -threads=12 -alloc_mode=3 -png=snowman.png
Case 2: --bind-to socket
mpirun -n 4 --bind-to socket /raytracer -width=512 -height=512 -spp=128 -threads=48 -alloc_mode=3 -png=snowman.png
Questions: - What is difference between Case 1 and Case 2. Any difference in performance? How many workers? - How could you adjust process/thread counts to better utilize the hardware in Case 2?
- MPI and thread pinning is hardware-aware.
- If the number of processes matches the number of domains (socket or NUMA), then the number of threads should equal the cores per domain to fully utilize the node.
- No speedup in Case 2: Oversubscription occurs because we requested 4 processes on a system with only 2 sockets.
- Threads compete for the same cores → OpenMPI queues threads and waits until other processes finish.
Best Practices for MPI Process and Thread Pinning
Mapping vs. Binding Analogy
Think of running MPI processes and threads like booking seats for a group of friends:
-
Mapping is like planning where your group will sit
in the theatre or on a flight.
- Example: You decide some friends sit in Economy, some in Premium
Economy, and some in Business.
- Similarly,
--map-bydistributes MPI ranks across nodes, sockets, or NUMA regions.
- Example: You decide some friends sit in Economy, some in Premium
Economy, and some in Business.
-
Binding is like reserving the exact seats for each
friend in the planned area.
- Example: Once the seating area is chosen, you assign specific seat
numbers to each friend.
- Similarly,
--bind-topins each MPI process or thread to a specific core or hardware unit to avoid movement.
- Example: Once the seating area is chosen, you assign specific seat
numbers to each friend.
This analogy helps illustrate why mapping defines placement and binding enforces it.
We will use --bind-to core (the smallest hardware unit)
and --map-by to distribute MPI processes across sockets or
NUMA or node regions efficiently.
Choosing the Smallest Hardware Unit
Binding processes to the smallest unit (core) is recommended because:
Exclusive use of resources
Each process or thread is pinned to its own core, preventing multiple threads or processes from competing for the same CPU.Predictable performance
When processes share cores, execution times can fluctuate due to scheduling conflicts. Binding to cores ensures consistent timing across runs.
- Best practice: Always bind processes to the smallest unit (core) and
spread processes evenly across the available hardware using
--map-by. - Example options:
-
--bind-to core→ binds each process to a dedicated core (avoids oversubscription).
-
--map-by socket:PE=<threads>→ spreads given number of threads as a processing element across the socket -
--map-by numa:PE=<threads>→ spreads processes across NUMA domains, assigning<threads>cores per process. - similarly
--map-by numa:PE=<threads> -
--cpus-per-rank <n>→ Assigns<n>cores (hardware threads) to each MPI rank - ensuring that all threads within a rank occupy separate cores.
-
Exercise
Use the given best practices above for case 1: -n 8,
-threads=1 and case 2: -n 8,
-threads=4 and answer following questions
Questions: - How many cores does the both jobs use? - Did you get more workers than you requested? - Did you see the scaling when running with 4 threads?
- 8 and 32
- No.
- Yes
Summary
Always check how pinning works
Use verbose reporting (e.g.,--report-bindings) to see how MPI processes and threads are mapped to cores and sockets.Documentation is your friend
For OpenMPI withmpirun, consult the manual: https://www.open-mpi.org/doc/v4.1/man1/mpirun.1.phpKnow your hardware
Understanding the number of sockets, cores per socket, and NUMA regions on your cluster helps you make effective binding decisions.Avoid oversubscription
Assigning more threads or processes than available cores hurts performance — it causes contention and idle waits.Recommended practice for OpenMPI
Use--bind-to corealong with--map-byto control placement and threads per process to maximize throughput.
Content from How to identify a bottleneck?
Last updated on 2025-09-24 | Edit this page
Overview
Questions
- How can I find the bottlenecks in a given job?
- What are common workflows to evaluate performance?
- What are some common types of bottlenecks?
Objectives
After completing this episode, participants should be able to …
- Choose between multiple workflows to evaluate job performance.
- Name typical performance issues.
- Determine if their job is affected by one of these issues.
How to identify a bottleneck?
Summary
Leading question: We were looking at a standard configuration with CPU, Memory, Disks, Network, so far. What about GPU applications, which are very common these days?
- General advice on the workflow
- Performance reports may provide an automated summary with recommendations
- Performance metrics can be categorized by the underlying hardware, e.g. CPU, memory, I/O, accelerators.
- Bottlenecks can appear by metrics being saturated at the physical limits of the hardware or indirectly by other metrics being far from what the physical limits are.
- Interpreting bottlenecks is closely related to what the application is supposed to do.
- Relative measurements (baseline vs. change)
- system is quiescent, fixed CPU freq + affinity, warmups, …
- Reproducibility -> link to git course?
- Scanning results for smoking guns
- Any best practices etc.
Content from Performance of Accelerators
Last updated on 2025-09-24 | Edit this page
Overview
Questions
- What are accelerators?
- How do they affect my jobs performance?
- How can I measure accelerator utilization?
Objectives
After completing this episode, participants should be able to …
- Understand difference of performance measurements on accelerators (GPUs, FPGAs) to CPUs.
- Understand how batch systems and performance measurements tools treat accelerators.
Introduction
Run the same example workload on GPU and compare.
Summary
Leading question: Performance optimization is a deep topic and we are not done learning. How could I continue exploring the topic?
- Tools to measure GPU/FPGA performance of a job
- Common symptoms of GPU/FPGA problems
Content from Next Steps
Last updated on 2025-10-29 | Edit this page
Overview
Questions
- What are other patterns of performance bottlenecks?
- How to evaluate an application in more detail?
Objectives
After completing this episode, participants should be able to …
- Find collection of performance patterns on hpc-wiki.info
- Identify next steps to take with regard to performance optimization.
Next Steps
hpc-wiki.info - I/O - CPU Front End - CPU Back End - Memory leak - Oversubscription - Underutilization
Summary
- There are many profilers, some are language-specific, others are vendor-related, …
- Simple profile with exclusive resources
- Repeated measurements for reliability